Teori om valensbindinger var den første av de kvantemekaniske teoriene som ble brukt for å tilnærme naturen til kjemiske bindinger i komplekse forbindelser. Søknaden var basert på ideen om giver-akseptor mekanisme dannelse av kovalente bindinger mellom liganden og kompleksdanneren. ligand teller donorpartikkel i stand til å overføre et par elektroner akseptor – kompleksdannende middel, som gir frie kvanteceller (atomorbitaler) av deres energinivåer for bindingsdannelse.
For dannelse av kovalente bindinger mellom kompleksdannende middel og ligander, er det nødvendig at ledig s-, s- eller d-atomiske orbitaler av kompleksdannende middel har gjennomgått hybridisering en bestemt type. Hybride orbitaler opptar en viss posisjon i rommet, og antallet deres tilsvarer koordinasjonsnummer kompleksdannende middel.
Dette skjer ofte assosiasjon av uparrede elektroner kompleksdannende middel i par, som tillater frigjøring av et visst antall kvanteceller - atomorbitaler, som deretter deltar i hybridisering og dannelse av kjemiske bindinger.
De ensomme elektronparene til liganden samhandler med hybridorbitalene til det kompleksdannende middelet, og overlapp korresponderende orbitaler av kompleksdannende middel og ligand med tilsynekomsten av økt elektrontetthet i det indre nukleære rommet. Elektronparene til det kompleksdannende middelet samhandler på sin side med de ledige atomorbitalene til liganden, styrking av forbindelsen ved hjelp av dativmekanismen. Dermed er den kjemiske bindingen i komplekse forbindelser den vanlige kovalent tilstrekkelig tilkobling varig Og energisk lønnsomt.
Elektronpar lokalisert i hybridorbitalene til kompleksdannende middel har en tendens til å innta en slik posisjon i rommet der deres gjensidige frastøtning vil være minimal. Dette leder til struktur komplekse ioner og molekyler viser seg å være i en viss avhengighet av type hybridisering.
La oss vurdere dannelsen av noen komplekser fra synspunktet til teorien om valensbindinger. Først og fremst bemerker vi at valensorbitalene til atomene til de kompleksdannende midlene er nære i energi:
E (n- 1)d » E ns » E np » E nd
|
Type hybridisering |
Kompleks geometri |
||
|
lineær |
-
|
||
|
trekantet |
- |
||
|
tetraeder |
2-
|
||
|
2-
|
|||
|
sp 3 d(z 2) |
trigonal bipyramide |
||
|
sp 3 d(x 2 - y 2) |
firkantet pyramide |
3-
|
|
|
sp 3 d 2 , |
3+ |
||
|
sp 3 d 3 |
femkantet bipyramide |
4-
|
For eksempel inkluderer 2+-kationen et sink(II)-kompleksdannende middel. Elektronskallet til dette betingede ionet har formelen 3 d 10 4s 0 4s 0 og kan betinget avbildes som følger:
Ledig 4 s- og 4 s-orbitaler av sink(II)-atomet danner fire sp 3-hybride orbitaler orientert mot toppene til tetraederet.
Hvert ammoniakkmolekyl har et ensomt elektronpar ved nitrogenatomet. Orbitaler av nitrogenatomer som inneholder ensomme elektronpar overlapper med sp 3-hybride orbitaler av sink(II), som danner et tetraedrisk komplekskation av tetraamminzink(II)2+: 
Siden det ikke er noen uparrede elektroner i 2+-ionet, viser det seg diamagnetisk egenskaper.
Tetrakloromanganat (II) -ion 2- inneholder fem uparrede elektroner per 3 d-orbitaler og ledige 4 s- og 4 s-orbitaler. Det dannes ledige orbitaler sp 3 hybrid orbitaler som overlapper med s-atomorbitaler av kloridioner: 
Det således oppnådde tetraedriske ion 2- er paramagnetisk, siden den inneholder fem uparrede elektroner.
Bruker den vanlige prediksjonsalgoritmen type hybridisering av atomorbitaler innenfor rammen av metoden for valensbindinger, er det mulig å bestemme geometri av komplekser annen sammensetning. For å gjøre dette, først av alt, er det nødvendig å skrive en elektronisk formel for valensnivået og konstruere et skjema for fordeling av elektroner over kvanteceller. For eksempel, for et nøytralt nikkelatom:

Overgang 4 s-elektroner per 3 d-undernivå transformerer paramagnetisk Ni-atom 0 tommer diamagnetisk partikkel Ni*: 
De resulterende ledige orbitalene gjennomgår hybridisering, og danner en tetraedrisk konfigurasjon. Så bygget tetraedrisk diamagnetisk tetrakarbonylnikkelkompleks (CN = 4), som er preget av betydelig stabilitet.
Hvis kompleksdanneren er nikkel(II) med den elektroniske konfigurasjonen 3 d 8 4s 0 4s 0, så behovet for å flytte elektroner fra 4 s-subnivå før hybridisering forsvinner, siden det er et tilstrekkelig antall ledige orbitaler til å implementere koordinasjonsnummer 4: 
En slik struktur har en ustabil paramagnetisk kompleks tetrabromikkolat(II)-ion 2-. Men når du kombinerer to elektroner 3 d-subnivå til et par og transformasjon av en av kvantecellene til dette undernivået til en ledig, både typen hybridisering og egenskapene til den resulterende komplekse endringen: 
Type hybridisering dsp 2 og den kvadratiske plane formen av komplekset realiseres ved dannelsen av en stall diamagnetisk kompleks av tetracyanoniccolate(II)-ion 2- (CN = 4): 
Hvis syntesen av cyanidkomplekset utføres under forhold med overskudd av ligand, kan koordinasjonsnummeret 5 realiseres: 
Stabil diamagnetisk det komplekse pentacyanoniccolate(II)-ion 3- har formen av en firkantet pyramide: 
Det oktaedriske nikkel(II) 2+-komplekset, men paramagnetisk men ganske stabil. Utdanningen hans er på grunn sp 3 d 2-hybridisering av nikkel-atomorbitaler: 
Hvis atomorbitaler av den ytre d-undernivå, komplekset er vanligvis stort sett paramagnetisk og ringte ytre bane eller høyspinn. Strukturen til slike komplekser kan tilsvare typen hybridisering, for eksempel, sp 3 d 2 .
Slike komplekser, under dannelsen av hvilke hybridisering finner sted med deltakelse av atomorbitaler til de preeksterne d-undernivåer kalles intraorbital eller lavt spinn og vanligvis diamagnetisk eller svakt paramagnetisk(alle eller nesten alle elektronene i det kompleksdannende middelet er sammenkoblet, og typen hybridisering, for eksempel, d 2 sp 3 eller dsp 2).
Undersøkelse av jern(II)-komplekser avslører både ytre-orbitale og intra-orbitale komplekser.


Diagrammet nedenfor viser hvordan paramagnetisk høyspinn heksafluorferrat(II)-ion 4- og diamagnetisk lavt spinn heksacyanoferrat(II)-ion 4-.
I seg selv svarer teorien om valensbindinger ikke på spørsmålet om hvilken type kompleks som dannes i hvert enkelt tilfelle, siden denne metoden ikke tar hensyn til påvirkningen av ligandens natur. Derfor må metoden for valensbindinger nødvendigvis suppleres med data om de magnetiske egenskapene til komplekset eller informasjon om effekten av liganden på naturen til det resulterende komplekset.
.Krystallfeltteori kom til å erstatte teorien om valensbindinger på 40-tallet av XX-tallet. I sin rene form brukes den ikke for tiden, siden den ikke kan forklare dannelsen av kovalente bindinger i komplekse forbindelser og ikke tar hensyn til den sanne tilstanden til ligander (for eksempel deres faktiske størrelser) selv i tilfelle av interaksjoner nær rent elektrostatisk.
Siden midten av 1950-tallet har den forenklede teorien om krystallfeltet blitt erstattet av en forbedret. ligandfeltteori, som tar hensyn til den kovalente naturen til de kjemiske bindingene mellom kompleksdanneren og liganden.
Imidlertid gir den mest generelle tilnærmingen til å forklare dannelsen av komplekse forbindelser molekylær orbital teori(MO), som for tiden råder over alle andre. Den molekylære orbitalmetoden sørger for både ren elektrostatisk interaksjon i fravær av overlappende atomorbitaler, og hele settet med mellomliggende grader av overlapping.
Vurder de grunnleggende konseptene krystallfeltteori, som i likhet med teorien om valensbindinger fortsatt beholder sin betydning for den kvalitative beskrivelsen av kjemiske bindinger i komplekse forbindelser på grunn av sin store enkelhet og klarhet.
I krystallfeltteorien vurderes det kjemiske bindingskompleksdannende middel - ligand elektrostatisk. I samsvar med denne teorien er ligander lokalisert rundt det kompleksdannende middelet ved toppunktene til vanlige polyedre ( polyedre) som punktgebyrer. Det virkelige volumet av liganden er ikke tatt i betraktning av teorien.
Ligander, som punktladninger, skaper rundt kompleksdannende middel elektrostatisk felt("krystallfelt", hvis vi vurderer en krystall av en kompleks forbindelse, eller ligandfelt), der energinivåene til kompleksdanneren og fremfor alt, d-undernivåer dele, og energien deres endres. Naturen til splitting, energien til nye energinivåer avhenger av symmetri arrangement av ligander (oktaedrisk, tetraedrisk eller annet krystallfelt). Når H 2 O, NH 3 , CO og andre molekyler er koordinert som ligander, anses de som dipoler orientert med en negativ ladning til kompleksdanneren.
La oss vurdere tilfellet med et oktaedrisk arrangement av ligander (for eksempel 3- eller 3+). I midten av oktaederet er det et atom som kompleksbinder M (+ n) med elektroner på d-atomiske orbitaler, og på toppene - ligander i form av punkt negative ladninger (for eksempel F-ioner - eller polare molekyler som NH 3). I et betinget M(+n)-ion som ikke er assosiert med ligander, er energiene til alle fem d-AO er de samme (dvs. atomorbitaler degenerert).
Imidlertid i det oktaedriske feltet av ligander d-AO kompleksdannende middel komme inn ulik posisjon. atomorbitaler d(z 2) og d(x 2 -
y 2), langstrakt langs koordinataksene, er nærmest ligandene. Mellom disse orbitalene og ligander lokalisert ved toppunktene til oktaederet, betydelig frastøtende krefter fører til en økning i energien til orbitalene. Disse atomorbitalene er med andre ord underlagt maksimal effekt av ligandfeltet. En sterkt komprimert fjær kan tjene som en fysisk modell av en slik interaksjon. 

Tre andre d-AO - d(xy), d(xz) Og d(yz) plassert mellom koordinataksene og mellom ligandene er plassert i større avstand fra dem. Samspill av slike d-AO med ligander er minimal, og derav energien d(xy), d(xz) Og d(yz)-AO synker sammenlignet med startverdien. 

Dermed femdoblet degenerert d-AO kompleksdannende middel, kommer inn oktaedrisk ligandfelt, blir utsatt for splitting i to grupper av nye orbitaler - tre ganger degenererte orbitaler med lavere energi d(xy), d(xz) Og d(yz), Og dobbelt degenererte orbitaler med høyere energi d(z 2) og d(x 2 -
y 2). Disse nye gruppene d-orbitaler med Nedre Og høyere energi utpeke d e og d g: 
Energiforskjell to nye undernivåer d e og d g ble navngitt deleparameter D0:
E 2 – E 1 = D0
Plassering av to nye energi subnivåer d e og d g i forhold til originalen ( d-AO) på energidiagrammet asymmetrisk:
(E 2 – E 0) > (E 0 – E 1).
Kvantemekanisk teori krever at med hele populasjonen av nye energinivåer av elektroner, forble den totale energien uendret, dvs. hun burde bli lik E 0 .
Med andre ord, likestillingen
4(E 2 – E 0) = 6(E 0 – E 1),
hvor 4 og 6 er maksimum antall elektroner pr d g-og d e-AO. Av denne likestillingen følger det at
(E 2 – E 0) / (E 0 – E 1) = 3/2 og
(E 2 – E 1) / (E 0 – E 1 >) = 5/2, eller
D 0 / ( E 0 – E 1) = 5/2, hvorfra ( E 0 – E 1) = 2/5 ´ D 0 >. Plassering av hvert elektron av de seks maksimalt mulig på d e-orbitaler årsaker avta (vinne) energi med 2/5 D 0 . Motsatt er plasseringen av hver av de fire mulige elektronene på d g -orbitaler forårsaker øke (koste) energi med 3/5 D 0 . Hvis befolket med elektroner d e-og d g orbitaler helt, så nei vinne energi vil ikke(som det ikke vil ekstra energikostnad): 4 ´ 3/5 ´ D 0 - 6 ´ 2/5 ´ D 0 = 0. Men hvis originalen d-AO bare bebodd delvis og inneholder fra 1 til 6 elektroner, og disse elektronene plasseres kun på d e -AO, så får vi betydelig energigevinst. Spesifisiteten til hver av liganden påvirker hvilket felt denne liganden skaper - sterk eller svak. Hvordan sterkere felt ligander enn mer betydning deleparameter D0. Studiet av splittingsparameteren er vanligvis basert på spektroskopisk forskning. Bølgelengder absorpsjonsbånd komplekser l i krystallinsk tilstand eller i løsning, på grunn av overgangen av elektroner fra d e - på d g-AO er assosiert med deleparameter D0 som følger: n = 1/l; D hvor er Plancks konstant h lik 6.626 ´ 10 - 34 J. s; Delingsparameter, i tillegg til typen ligand, avhenger på graden av oksidasjon Og natur kompleksdannende middel. På økning i atomladning kompleksdannende atom D 0 øker også. Kationer av heksamminekobolt(III) 3+ , heksamminerhodium(III) 3+ , heksammineridium(III) 3+ ( Z= 27, 45 og 77) er karakterisert ved å dele parametere lik 22900, 34100 og 41000 cm-1. Avhengigheten av D0 av arten av liganden er mer variert. Som et resultat av studiet av en rekke komplekse forbindelser, ble det funnet at, i henhold til evnen til å øke spaltningsparameteren til kompleksdannende metaller som er i deres vanlige oksidasjonstilstander, kan de vanligste ligander ordnes i følgende spektrokjemisk serie, langs hvilken verdien av D 0 vokser monotont: Dermed det sterkeste elektrostatiske feltet rundt kompleksdanneren og den sterkeste spaltningen d-AO er forårsaket av NO 2 - ligander, CN -
og CO. Vurder fordelingen av elektroner over d e-og d g orbitaler i det oktaedriske feltet av ligander. Bosetting d e-og d g -orbitaler oppstår i full overensstemmelse med Gunds styre Og Pauli-prinsippet. I dette tilfellet, uavhengig av verdien av splitteparameteren, okkuperer de tre første elektronene kvanteceller d e-undernivå: Hvis antall elektroner i d- det er mer enn tre undernivåer av kompleksdanneren, det er to muligheter for å plassere dem på delte undernivåer. Ved en lav verdi av spaltningsparameteren (svak ligandfelt) overvinner elektronene energibarrieren. d e-og d g-orbitaler; det fjerde og deretter det femte elektronet fyller kvantecellene d g-undernivå. Med et sterkt felt av ligander og en høy verdi på D 0 er populasjonen av det fjerde og femte elektronet d g -undernivå ekskludert; utfylling pågår d e orbitaler. På svakt ligandfelt populerer kvanteceller 4 eller 5 elektroner har parallelle spinn, så det resulterende komplekset viser seg å være sterkt paramagnetisk. I et sterkt ligandfelt ett og deretter to elektronpar dannes d e -undernivå, altså paramagnetisme komplekset er mye svakere. Det sjette, syvende og åttende elektronet i tilfelle av et svakt felt er igjen på d e -undernivå, som komplementerer konfigurasjonene til elektronpar (ett i tilfellet d 6, to - d 7 og tre - d 8): Ved et sterkt ligandfelt fyller det sjette elektronet seg d e -AO, som fører til diamagnetisme kompleks, hvoretter det syvende og åttende elektron kommer inn i d g-undernivå: Åpenbart med en åtte-elektronkonfigurasjon strukturelle forskjeller mellom komplekser med ligander svak Og sterkt felt forsvinner. Okkupasjonen av orbitaler av det niende og tiende elektronet er heller ikke forskjellig for komplekser av begge typer: La oss gå tilbake til betraktningen av den elektroniske strukturen til oktaedriske komplekse ioner 3+ og 3-. I henhold til beliggenhet i spektrokjemisk serie, er ammoniakk NH3 en av ligandene sterkt felt
, og fluoridionet F - - svakt felt
. Følgelig vil populasjonen av atomorbitaler i disse kompleksene med elektroner oppstå i henhold til skjemaet: I anion 3 - ligander F - skaper et svakt krystallfelt (D 0 = 13000 cm - 1), og alle elektronene til de opprinnelige 3 d 6 -JSC er plassert på d e-og d g orbitaler uten noen paring. Det komplekse ionet er høyspinn og inneholder fire uparrede elektroner, så det paramagnetisk. I 3+-ionet skaper NH 3-ligander et sterkt krystallfelt (D 0 = 22900 cm - 1), alle 3 d 6-elektroner er plassert på en mer energisk gunstig d e-orbitaler. Overføring av elektroner fra d e - på d g-orbitaler umulig forfaller også høy energibarriere. Derfor er denne komplekse kationen lavt spinn, den inneholder ikke uparrede elektroner og diamagnetisk. Ordningene for distribusjon av elektroner langs orbitaler i et oktaedrisk felt for ioner 2+ og 4- kan presenteres på lignende måte: H 2 O-ligander skaper et svakt felt; utveksling av elektroner mellom d e-og d g -orbitaler forårsaker ikke vanskeligheter og derfor er antallet uparrede elektroner i det komplekse ion det samme som i det betingede Fe + II ion. Det resulterende akvakomplekset - høyt spinn, paramagnetisk. Mange komplekse forbindelser i krystallinsk tilstand og i vandig løsning utmerker seg med lyse farger. Således er en vandig løsning som inneholder 2+ kationer farget intenst blå, 3+ kationer gir løsningen en fiolett farge og 2+ kationer rød. Teorien om krystallfeltet gjør det mulig å forklare utseendet til en eller annen farge i komplekse forbindelser. Hvis lys sendes gjennom en løsning eller en krystallinsk prøve av et stoff synlig del av spekteret, da er i prinsippet tre varianter av den fysiske oppførselen til prøven mulig: ingen lysabsorpsjon hvilken som helst bølgelengde (stoffprøve fargeløs, selv om det kan ha absorpsjonsbånd i det ultrafiolette området av spekteret); total absorpsjon av lys over hele bølgelengdeområdet (prøven vil vises svart); endelig, lysabsorpsjon bare spesifikk bølgelengde(da vil prøven ha farge komplementær til absorbert smal del av spekteret). Dermed blir fargen på en løsning eller krystaller bestemt frekvens av absorpsjonsbånd synlig lys: Absorpsjonen av lyskvanter av komplekser (for eksempel de som har en oktaedrisk struktur) forklares av samspillet mellom lys og elektroner lokalisert på d e -undernivå, ledsaget av deres overgang til ledige orbitaler d g-undernivå. For eksempel, når lys føres gjennom en vandig løsning som inneholder heksakvatitan(III) 3+ kationer, finnes et lysabsorpsjonsbånd i det gulgrønne området av spekteret (20300 cm - 1, l » 500 nm). Dette skyldes overgangen til et enkelt elektron av kompleksdannende middel med d e -AO på d g-undernivå: Derfor får en løsning som inneholder 3+ en fiolett farge (komplementær til den absorberte gulgrønne). Vanadiumsaltløsning Cl 3 er grønn. Dette skyldes også de tilsvarende overgangene av elektroner når de absorberer en del av energien til lysstrålen. I grunntilstand, med den elektroniske konfigurasjonen av vanadium(III) 3 d 2, to uparrede elektroner okkuperer d e-undernivå: Det er alt to alternativer for overgang av to elektroner på d g-undernivå: enten både et elektron er okkupert d g -AO, eller bare en av dem. Alle andre elektronoverganger assosiert med en reduksjon i totalt spinn er forbudt. Hvis kompleksdanneren har en elektronisk konfigurasjon d 0 eller d 10 da elektronoverganger Med d e - på d g -undernivå eller omvendt umulig enten fordi mangel på elektroner, eller fordi mangel på ledige orbitaler. Derfor absorberer ikke løsninger av komplekser med slike kompleksdannende midler som Sc(III), Cu(I), Zn(II), Cd(II), etc. energi i den synlige delen av spekteret og virker fargeløs: Selektiviteten til lysabsorpsjon avhenger ikke bare av kompleksdannende middel Og dens oksidasjonstilstand, men også fra typer ligander. Når ligander på venstre side av den spektrokjemiske serien erstattes i den komplekse forbindelsen med ligander som skaper sterk elektrostatisk felt observert øke brøkdel av energien absorbert av elektroner fra transmittert lys og som en konsekvens, avta bølgelengden til det tilsvarende absorpsjonsbåndet. En vandig løsning som inneholder tetraaquacopper(II) 2+ kationer er således farget blå, og en løsning av tetraamminecopper(II) 2+ sulfat har en intens blå farge. ________________________ Gjenta: >>> applikasjoner
Får energi gjennom fortrinnsvis oppgjør elektroner d e-atomiske orbitaler kalles energi for stabilisering av komplekset ved hjelp av liganderfeltet.
lysets hastighet Med
= 3 ´ 10 10 cm/s.
Enhet D 0 - det samme som for bølgetallet n: cm - 1, som omtrent tilsvarer 12 J / mol.
I komplekse forbindelser som inneholder kompleksdannende midler av samme periode og i samme oksidasjonstilstand, med de samme ligander, er spaltningsparameteren omtrent den samme. Med en økning i graden av oksidasjon av kompleksdannende middel, verdien av D 0 øker. For akvakomplekser 2+ og 2+ er verdien av spaltningsparameteren 7800 og 10400 cm - 1, og for 3+ og 3+ - henholdsvis 13700 og 21000 cm - 1.
I - Br -Cl -»NCS- NEI 3 -F -ÅH -H2O » H-NH3 NR 2 - CN - » NEI » CO. 
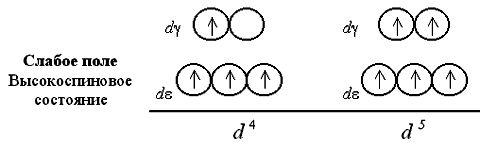








Omvendt forårsaker ligander CN - betydelig spaltning d-AO, som er 33000 cm - 1. Dette betyr at det er en sterk tendens til å romme alle elektroner på d e orbitaler. Energigevinst, oppnådd med en slik populasjon av orbitaler, er mye større enn energikostnadene på grunn av sammenkobling av elektroner.


Disse overgangene av elektroner som har mottatt overflødig energi tilsvarer absorpsjonsbånd ca. 400 nm i absorpsjonsspekteret til en hexaaquavanadium(III)kloridløsning. Absorpsjon i det lilla-fiolette området av spekteret gir en ekstra farge til løsningen - lysegrønn.

Skapt av omkringliggende ioner, atomer eller molekyler. Konseptet med et krystallfelt ble foreslått av Becquerel for å beskrive tilstanden til atomer i krystaller og deretter utviklet av Hans Bethe og John Van Vleck for å beskrive de laveste tilstandene av overgangsmetallkationer omgitt av ligander, både anioner og nøytrale molekyler. Krystallfeltteorien ble ytterligere kombinert [og forbedret] med teorien om (delokaliserte) molekylære orbitaler til en mer generell ligandfeltteori som tar hensyn til den partielle kovalensen til metall-ligandbindingen i koordinasjonsforbindelser.
Krystallfeltteori gjør det mulig å forutsi eller tolke de optiske absorpsjonsspektrene og spektrene til elektronisk paramagnetisk resonans av krystaller og komplekse forbindelser, så vel som entalpiene for hydratisering og stabilitet i løsninger av overgangsmetallkomplekser.
Encyklopedisk YouTube
-
1 / 5
I følge TCP oppstår interaksjonen mellom et overgangsmetall og ligander på grunn av tiltrekningen mellom et positivt ladet metallkation og en negativ ladning av elektroner i ikke-bindende ligandorbitaler. Teorien vurderer endringen i energien til fem degenerert d-orbitaler omgitt av punktladninger av ligander. Når liganden nærmer seg metallionet, kommer ligandelektronene nærmere noen d-orbitaler enn til andre, noe som forårsaker tap av degenerasjon. Elektroner d-orbitaler og ligander frastøter hverandre som ladninger med samme fortegn. Dermed energien til de d-elektroner som er nærmere ligandene blir høyere enn de som er lenger unna, noe som resulterer i en splittelse av energinivåene d-orbitaler.
Splitting påvirkes av følgende faktorer:
- Naturen til metallionet.
- Graden av oksidasjon av metallet. Jo høyere oksidasjonstilstand, jo høyere er spaltningsenergien.
- Plassering av ligander rundt et metallion.
- Naturen til liganden som omgir metallionet. Jo sterkere effekten av liganden er, desto større er forskjellen mellom høye og lave energinivåer.
Den vanligste formen for ligandkoordinering er oktaedrisk, hvor seks ligander skaper et krystallfelt med oktaedrisk symmetri rundt metallionet. I det oktaedriske miljøet til et metallion med ett elektron i det ytre skallet, er d-orbitalene delt inn i to grupper med forskjell i energinivåer Δ okt ( dele energi), mens energien til orbitalene dxy, dxz Og d yz vil være lavere enn d z 2 og d x 2 -y 2, siden orbitalene til den første gruppen er lenger fra liganden og opplever mindre frastøting. De tre lavenergiorbitalene er betegnet som t2g, og to med høy like f.eks.
De nest vanligste er tetraedrisk komplekser der fire ligander danner et tetraeder rundt et metallion. I dette tilfellet d-orbitaler er også delt inn i to grupper med forskjell i energinivå Δ tetra. I motsetning til oktaedrisk koordinasjon vil orbitaler ha lav energi d z 2 og d x 2 -y 2, og høy - d xy , d xz Og d yz. I tillegg, siden elektronene til liganden ikke er direkte i retningen d-orbitaler, vil spaltningsenergien være lavere enn ved oktaedrisk koordinasjon. Ved hjelp av TST kan man også beskrive flat firkant og andre komplekse geometrier.
Energinivåforskjellen Δ mellom to eller flere grupper av orbitaler avhenger også av arten av liganden. Noen ligander forårsaker mindre spaltning enn andre, noe som forklares med ligandfeltteori. Spektrokjemisk serie- empirisk oppnådd liste over ligander, ordnet i stigende rekkefølge Δ:
Oksydasjonstilstanden til metallet påvirker også Δ. Et metall med høyere oksidasjonstilstand tiltrekker ligander nærmere på grunn av den større ladningsforskjellen. Ligander nærmere metallionet forårsaker mer spaltning.
Lav- og høyspinnkomplekser
Store spaltningsligander d-nivåer, for eksempel CN - og CO, kalles ligander sterkt felt. I komplekser med slike ligander er det ugunstig for elektroner å okkupere høyenergiorbitaler. Derfor er lavenergiorbitalene fullstendig fylt før fyllingen av høyenergiorbitalene begynner. Slike komplekser kalles lavt spinn. For eksempel er NO 2 - en sterk feltligand som skaper en stor splittelse. Alle 5 d-elektronene til det oktaedriske ionet 3− vil være plassert på lavere nivå t 2g .
I motsetning til dette kalles ligander som forårsaker liten splitting, slik som I − og Br − , ligander svakt felt. I dette tilfellet er det lettere å sette elektroner i høyenergibaner enn det er å sette to elektroner i samme lavenergibane, fordi to elektroner i en bane frastøter hverandre, og energikostnadene ved å plassere et andre elektron i en bane er høyere enn Δ. Således, før sammenkoblede elektroner dukker opp, i hver av de fem d-orbitaler må plasseres ett elektron om gangen i henhold til Hunds regel. Slike komplekser kalles høyspinn. For eksempel er Br − en svak feltligand som forårsaker en liten splittelse. Alle 5 d-orbitaler til 3−-ionet, som også har 5 d-elektroner vil være okkupert av ett elektron.
Splittingsenergien for tetraedriske komplekser Δ tetra er omtrent lik 4/9Δ okt (for samme metall og ligander). Som et resultat, energinivåforskjellen d-orbitaler er vanligvis under elektronparingsenergien, og tetraedriske komplekser er vanligvis høyspinn.
Distribusjonsdiagrammer d-elektroner gjør det mulig å forutsi de magnetiske egenskapene til koordinasjonsforbindelser. Komplekser med uparrede elektroner er paramagnetiske og tiltrekkes av et magnetfelt, mens komplekser uten dem er diamagnetiske og frastøter hverandre svakt.
Krystallfeltteori (CFT) er en utvikling av en enkel elektrostatisk teori om kompleks dannelse. Det er best brukt på tilkoblinger d- elementer og er den enkleste modellen som lar deg enkelt forklare egenskapene deres. I følge teorien
bindingen i komplekset utføres på grunn av den elektrostatiske interaksjonen mellom det positivt ladede sentralatomet og de negativt ladede ligandene. Liganden betraktes kun som en ladningskilde (krystallfelt), mens det for sentralatomet tas hensyn til det romlige arrangementet av d-orbitaler .
Opprinnelig ble TQP brukt for å forklare egenskapene til krystallinske stoffer og fikk derfor navnet sitt. Imidlertid er den like anvendelig for alle systemer med geometrisk regelmessig anordnede elektrisk samvirkende partikler, for eksempel til et enkelt komplekst ion.
Den geometriske strukturen til en kompleks partikkel bestemmes i den første tilnærmingen av maksimal gjensidig frastøtning av negativt ladede ligander: seks ligander danner et oktaeder, fire - et tetraeder.
I et fritt atom eller ion, alle fem d- orbitaler på samme nivå har samme energi, dvs. de er degenererte. Hvis, hypotetisk, et ion d- element til midten av sfæren av en likt fordelt negativ ladning, vil den samme frastøtende kraften virke på alle fem elektronskyene. Dette vil resultere i opphisselse d- undernivå, men degenerasjonen er ikke opphevet. Et annet bilde oppstår hvis ionet kommer inn i et oktaedrisk, tetraedrisk eller annet miljø (mindre symmetrisk enn sfærisk). La oss si et positivt ion d- elementet er i et oktaedrisk miljø av negativt ladede ioner eller polare molekyler.
I dette tilfellet opplever - og - elektronene en større elektrostatisk frastøtning fra liganden enn d xy -, d xz - Og d yz - elektroner (Figur 2.5).
Derfor energien d- elektroner under disse forholdene er ikke det samme: i - og - tilstand () er energien høyere enn i d xy -, d xz - Og d yz - stat(). Således, hvis i et fritt ion eller i et sfærisk felt er det fem d- orbitaler har samme energi, så i det oktaedriske feltet av ligander er de delt inn i to grupper med forskjellige energier - i tre og to orbitaler.
Energiforskjell d- nivåer D kalles dele energi av krystallfeltet . Det uttrykkes i enheter Dq(et mål på styrken til krystallfeltet), og D E = E 1 - E 2 = 10Dq= E. For et oktaedrisk kompleks, energien til -orbitaler per 2/5D (4 Dq) under degenerert d- orbitaler, ikke sant? for 3/5D (6 Dq) høyere.
Verdien av spaltningsenergien bestemmer egenskapene til CS, så det er viktig å vite hvilke faktorer den avhenger av.
1. Type koordinering av sentralatomet.
D-parameteren påvirkes både av antall ligander som omgir CA og av deres gjensidige arrangement. Spaltningsenergien med et oktaedrisk felt av ligander (D o), alt annet like, er alltid høyere enn ved et tetraedrisk felt (D t):
D t = D O . (2)
Dette forklares av den forskjellige størrelsen på den elektrostatiske interaksjonen mellom CA-elektronene og liganden (se fig. 2.8).
2. Ladning av det sentrale ion.
Jo høyere ladning det sentrale ion har, desto sterkere er dets elektrostatiske interaksjon med ligander og jo større er spaltningsenergien. Når du øker ladningen fra +2 til +3 for de fleste 3 d- elementer, øker spaltningsenergien omtrent 1,5 ganger (tabell 2.2).
Tabell 2.2.
3. Elektronisk struktur av det sentrale ion
Splitte energi i komplekser 4 d- elementer med omtrent 50 %, og i komplekser 5 d- elementer er 75 % høyere enn i de tilsvarende metallkompleksene 3 d- rad. Dette skyldes de forskjellige lengdene på orbitalene i rommet.
4. Ligandens natur
I henhold til graden av økning i spaltningsparameteren D, er ligandene ordnet i en rad kalt spektrokjemisk (Fig. 2.9).
Ris. 2.9. Spektrokjemisk serie av ligander
I samspillet mellom en sterk feltligand og CA oppstår splitting d- orbitaler (avsnitt 2.3, fig. 2.6). I dette tilfellet blir fordelingen av elektroner i henhold til Hunds regel umulig, siden overgangen av elektroner fra et lavere -nivå til et høyere -nivå krever energi, noe som er energetisk ugunstig (en stor verdi av splitteparameteren D). Derfor fyller elektronene først -nivået helt, og deretter fylles bare -nivået. I tilfelle å være på d- orbitaler på 6 elektroner, under påvirkning av en sterk feltligand, er -nivået fylt med sammenkobling av elektroner. Dette skaper lavt spinn diamagnetisk kompleks. Og i tilfellet med en svak feltligand, når splittelsesparameteren D får en lavere verdi, blir en jevn fordeling av elektroner i henhold til Hund-regelen mulig. I dette tilfellet skjer ikke sammenkoblingen av alle elektroner; høyspinn paramagnetisk kompleks.
Sekvensen for arrangement av ligander i den spektrokjemiske serien innenfor rammen av MO-teori kan forklares som følger. Jo større grad av overlapping av de innledende orbitalene er, desto større energiforskjell mellom bindings- og løsneorbitalene og jo større D. Med andre ord øker verdien av D med økende y- metall-ligand-binding. Verdien av D er i tillegg betydelig påvirket av p-binding mellom CA og ligander.
Hvis ligander har orbitaler (tomme eller fylte) som, i henhold til symmetriforhold, er i stand til å overlappe med d xy -, d xz - Og d yz - CA-orbitaler, så blir MO-diagrammet av komplekset mye mer komplisert. I dette tilfellet til MO y- Og på * - type, molekylære orbitaler p legges til - og p* - type. Orbitaler av ligander i stand til - overlappende, for eksempel p- Og d- atomorbitaler eller molekylær p - og p* - orbitaler av binukleære molekyler. På fig. 2.10 viser kombinasjoner av ligandorbitaler og d xz - CA orbital, som i henhold til symmetriforholdene kan kombineres for å danne molekylær p - orbitaler.

Ris. 2.10. d xz - CA orbital (a) og kombinasjoner som tilsvarer den i symmetri p-(b) og p * - (c) ligandorbitaler som fører til dannelsen av MO av det oktaedriske komplekset

Ris. 2.11. Påvirkning s - bindende for verdien av D
Deltakelse d xy -, d xz - Og d yz - orbitaler i p-konstruksjon - orbitaler fører til en endring i D. Avhengig av forholdet mellom energinivåene til CA-orbitalene og ligandorbitalene kombinert med dem, kan verdien av D øke eller redusere (fig. 2.11).
Når r dannes - orbitaler av komplekset, en del av elektrontettheten til CA overføres til liganden. En slik s - interaksjon kalles dativ. Når r dannes * - orbitaler av komplekset, blir en del av elektrontettheten overført fra liganden til CA. I så fall r - interaksjonen kalles donor-akseptor.
Ligander som er p - akseptorer forårsaker mer spaltning d- nivå; ligander som er p - givere, tvert imot, forårsaker en liten splittelse d- nivå. Naturen y- Og R- ligandinteraksjoner kan deles inn i følgende grupper.

Det foregående forklarer rekkefølgen av arrangement av ligander i den spektrokjemiske serien:
På begynnelsen av denne raden er de sterke feltligander, og på slutten er de svake feltligander.
Bond styrke
Denne verdien korrelerer med krystallfeltstabiliseringsenergi (ESKP) - en gevinst i energi på grunn av fylling av lav energi d- nivåer relativt udelte d- orbitaler. Når det gjelder kompleks 3? stabiliseringsenergien er lik forskjellen mellom forsterkningen på grunn av elektroner lokalisert i -orbitaler (2/5D 4) og tapet på grunn av elektroner i -orbitaler (3/5D 2): 3/5D o 2 = 2/5D o (eller 4 Dq). For lavspinnkomplekset 3+ vil stabiliseringsenergien være mye høyere, siden alle elektronene i den er i gunstige orbitaler (), men det bør tas i betraktning at under dannelsen av dette komplekset brukes energi også på elektronparing (2 R, siden den har to flere elektronpar enn i udelt tilstand): ESCP \u003d 2 / 5D o 6 - 2 R= 12/5 D o - 2 R(eller 24 Dq - 2R).
Magnetiske egenskaper
For komplekser 3 d- elementer, er det magnetiske momentet nær det som beregnes av formelen for den "rent spinnkomponenten":
m eff = , (3)
Hvor n- antall uparrede elektroner; m eff uttrykkes i Bohr-magnetoner (mB).
Tabell 2.3 presenterer verdiene til de eksperimentelle og beregnede verdiene for det magnetiske momentet for ulike komplekser.
Tabell 2.3. Magnetiske egenskaper til forskjellige komplekser

Farging av kompleksene
De fleste overgangselementkomplekser er fargede forbindelser, dvs. de er i stand til å absorbere energi i det synlige området av spekteret (bølgelengdeområde fra 410 til 720 nm, som tilsvarer energi fra 290 til 145 kJ/mol). Dette skyldes overgangen av elektroner fra et lavere til et høyere fri energinivå, som utføres på grunn av absorpsjon av synlig lyskvanter. I dette tilfellet absorberes lys med bølgelengden som tilsvarer splittende energi:

Den synlige fargen på forbindelsen tilsvarer ytterligere farge, dvs. fargen som vi ser hvis noen bølgelengder fjernes fra det kontinuerlige spekteret. På fig. 2.14 viser spekteret av titan (III) 3+ vannkomplekset. Fargen forklares av absorpsjonsspekteret til komplekset. Dette oktaedriske komplekset har en elektronisk konfigurasjon
Eksiteringen av et elektron c til en tilstand krever absorpsjon av et energikvante D:

Som det fremgår av fig. 2.14, tilsvarer dette absorpsjonen av stråling med en bølgelengde på 50 nm. Dermed overfører 3+ løsninger som absorberer gule stråler blått og rødt, så fargen på løsningene er fiolett.
Ved å bruke formel (4) kan vi utlede D-verdien for kompleks 3+, som er lik 238 kJ/mol.
Jahn-Teller-effekt
Til nå har kun oktaedriske og tetraedriske komplekser blitt vurdert. Imidlertid er det mange forbindelser med en forvrengt oktaedrisk struktur, kvadratiske komplekser og komplekser med andre koordinasjonstall, for eksempel 5, 7, etc. En betydelig forvrengning av den regulære oktaedriske strukturen er observert for komplekser med (Cr 2+ , Mn 3+) og (Cu 2+ , Ag 2+) konfigurasjoner. Dette kommer til uttrykk:
· i ulik bindingslengde mellom CA og ligander;
· i utvidelse eller bifurkasjon av linjer i absorpsjonsspektra.
I disse tilfellene inneholder de degenererte -orbitalene et oddetall elektroner, som kan være lokalisert i - eller -orbitaler. I følge Jahn-Teller-teoremet, hvis flere ekvivalente orbitalt degenererte energinivåer tilsvarer en tilstand av systemet, bør den geometriske forvrengningen av systemet fjerne orbitaldegenerasjonen og redusere den totale energien til systemet.
Siden kompleksdanneren i de fleste tilfeller er et metallkation, og ligandene er anioner eller høypolare molekyler, gir den elektrostatiske interaksjonen et betydelig bidrag til kompleksdannelsens energi. Det er dette krystallfeltteorien (CFT) fokuserer på. Navnet gjenspeiler det faktum at elektrostatisk interaksjon er karakteristisk først og fremst for krystaller av ioniske forbindelser.
Grunnleggende bestemmelser i teorien.
1. Bindingen mellom kompleksdannende middel og ligander betraktes som elektrostatisk.
2. Ligander anses å være punktioner eller punktdipoler, deres elektroniske struktur ignoreres.
3. Ligandene og det kompleksdannende middelet anses å være stivt fiksert.
4. Den elektroniske strukturen til kompleksdannende middel vurderes i detalj.
Vurder de vanligste oktaedriske kompleksene (fig. 4.1), analyser samspillet mellom ligander og de elektroniske orbitalene til sentralionet (fig. 4.2 og 4.3).
Ris. 4.1. Komplekserende ion i et oktaedrisk felt av ligander
Ris. 4.2. Interaksjon av ligander med s- og p-orbitaler i et oktaedrisk felt
Ris. 4.3. Interaksjon av ligander med d-orbitaler i et oktaedrisk felt
Som det fremgår av fig. 4,2 s- og p-orbitaler samhandler likt med ligander. Når det gjelder d-orbitaler, "ser" to av de fem direkte på liganden, mens de tre andre ser forbi dem (bare snittet ved zy-planet for d zy-orbitalen er vist i fig. 4.3; tilsvarende for d zx og d xy orbitaler). Orbitalene samhandler med andre ord sterkere med ligander enn orbitalene d zy , d zx , d xy . Følgelig, i det oktaedriske feltet av ligander, er fem orbitaler opprinnelig identiske i energi (de sier "fem ganger degenerert nivå") delt inn i to grupper: orbitalene vil ha en energi høyere enn orbitalene d zy , d zx og d xy (Fig. 4.4). Verdien av Δ okt kalles splittende energi og, innenfor rammen av TST, er energigevinsten som forårsaker dannelsen av elektronpar eller bevaring av den elektroniske tilstanden til sentralionet i komplekset.
Ris. 4.4. Splitting av d-nivået i det oktaedriske feltet av ligander
Hvis E-par > Δ okt, skjer ikke dannelsen av elektronpar og høyspinntilstanden bevares. Hvis Δ okt > E-par, så er det dannelse av elektronpar og en lavspinntilstand vil finne sted. Som allerede nevnt er dette kun mulig for Me 2+ cyanidkomplekser og for Me 3+ komplekser med ligander CN – , NO 2 – , NH 3 .
Hvis vi tar det samme sentrale ionet og bestemmer splittende energi for komplekset med forskjellige ligander, viser det seg at Δ okt øker i følgende sekvens, kalt den spektrokjemiske serien:
JEG-< Br – < Cl – < F – < OH – < H 2 O < NH 3 < NO 2 – < CN –
Den samme sekvensen er også bevart for kompleksene til det andre sentrale ion. Ligandene på venstre side av raden er svake feltligander, og liganden på høyre side av raden er høyfeltsligander. TST lar en finne en kvantitativ karakteristikk av gevinsten i bindingsenergi på grunn av elektrostatisk interaksjon - energien til stabilisering av et krystallfelt (ESF). Den totale energien til de fem d-orbitalene til et fritt ion er 5E d ; den er naturlig lik den totale energien til de fem delte orbitalene:
5 E d = 2 E Eg + 3 E T2 g
Til denne ligningen legger vi den åpenbare likheten:
E Eg – E T2 g = Δ okt
Løsningen av systemet med to gitte ligninger gir følgende resultater:
Så hvis et ion i et oktaedrisk felt har n elektroner på T 2 g orbitaler, og m elektroner på E g orbitaler:
For eksempel, for kompleksene 2+ og 4– diskutert ovenfor:
Svakt felt Sterkt felt
Det sterkere cyanidkomplekset har en betydelig høyere ESCR.
Splittingen av d-nivået til det sentrale atomet i det tetraedriske feltet av ligander fører til en reduksjon i energien til elektronene i orbitalene (disse orbitalene er rettet forbi liganden) og til dens økning i d xy , d xz og d yz orbitaler (rettet mot liganden), som vist i fig. 4.5.
Ris. 4.5. Spaltning av d-nivået i det tetraedriske komplekset
Splittingsenergien Δ tetra er mindre enn Δ okt; fra rent geometriske betraktninger følger det at Δ tetra = Δ okt. Åpenbart vil stabiliseringsenergien av krystallfeltet i dette tilfellet være:
TST gir en enkel forklaring på tilstedeværelsen eller fraværet av farge i et kompleks. Hvis elektroniske overganger mellom orbitalene T 2 g og E g er mulig (og dette er mulig med den elektroniske konfigurasjonen av sentralionet fra d 1 til d 9), blir de komplekse forbindelsene farget. Hvis slike overganger er umulige (og dette vil være tilfellet med de elektroniske konfigurasjonene til sentralionet d 0 eller d 10), er de komplekse forbindelsene fargeløse. Kompleksene av sølv, kobber (I), gull (I), sink, kadmium, kvikksølv (i alle tilfeller d 10), aluminium, magnesium, scandium, lantan (i alle tilfeller d 0) er fargeløse. Og kompleksene av kobber (II), gull (III) er allerede farget; fargede komplekse forbindelser av jern (II) og (III), nikkel, kobolt, etc. I disse tilfellene har de sentrale ionene en elektronisk konfigurasjon d n (n=1–9).
For å forklare den kjemiske bindingen i komplekse forbindelser, er ligandfeltteorien mye brukt, som ikke bare tar hensyn til den elektroniske strukturen til sentralionet (atomet), men også ligandene. I hovedsak skiller ikke ligandfeltteorien seg fra MO LCAO-metoden som er mye brukt i kvantekjemi.
En-elektronbølgefunksjonen til den molekylære orbitalen Ψ er representert som
Ψ = aΨ o + bΦ,
Φ=C 1 φ 1 + C 2 φ 2 + … + C i φ i ,
hvor Ψ o er atombanen til sentralionet (atomet); Φ er den molekylære orbitalen til ligandsystemet, φ i er den atomære eller molekylære orbitalen til den i-te liganden.
Teoretiske begreper viser at Ψ, Ψ o og Φ må ha samme symmetriegenskaper. Slike lineære kombinasjoner av atomorbitaler i ligandsystemet kalles "gruppeorbitaler".
Teorien om MO-metoden antyder at overlappingen av Ψ o og Φ orbitalene til en viss grad forekommer i alle tilfeller der dette tillates av symmetri. Som et resultat sørger denne teorien for både ren elektrostatisk interaksjon i fravær av orbital overlapping, og maksimal overlapping med et minimumsbidrag av den elektrostatiske komponenten av interaksjonen, og alle mellomliggende grader av overlapping.
Dermed er ligandfeltteorien den mest komplette og generelle teorien om kjemisk binding i komplekse forbindelser.
Krystallfeltteori (CFT)
- 4.2.1 Innledning. Dette er en forenklet teori om den elektroniske strukturen til d-elementkationer. Ordet "krystallinsk" i tittelen er uheldig. Teorien har ingenting med krystallografi å gjøre og er enda bedre anvendelig på isolerte grupper i løsning eller i en gass enn på krystaller. Men det er for sent å endre navn. Teorien beskriver strukturer med delvis fylt d-undernivå, men for sammenligning vil vi hele tiden involvere to ekstreme tilfeller - d 0 og d 10 , det vil si totalt 11 alternativer.
- 4.2.2 Hovedbestemmelser for TCH.
- 1. Forbindelsen av d-elementet kation med dets naboer - ligander - betraktes som en ren elektrostatisk tiltrekning, og ligander - som punkt negative ladninger - anioner eller negative ender av polare molekyler. Den elektroniske strukturen til liganden vurderes ikke. Dette er en veldig grov tilnærming, men overraskende effektiv. Selvfølgelig er bindingen i stor grad kovalent i naturen, og strukturen til liganden er veldig viktig. Men det tas implisitt i betraktning.
- 2. Under påvirkning av det elektrostatiske feltet til liganden deler d-subnivået seg. Fem d-orbitaler har samme energi i fravær av et eksternt felt eller i et sfærisk symmetrisk felt, og i feltet med ligander blir de ulik, energien til noen av dem reduseres, energien til andre øker, men deres sentrum av tyngdekraften på energiskalaen er bevart, altså gjennomsnitt energien til orbitalene forblir på samme nivå. Opplegg splitting (hvilke orbitaler senket energien, som økte) avhenger bare av formen på miljøet, men avhenger ikke av arten av kation og ligander, fordi de vinkelformede delene av bølgefunksjonene er de samme for alle elementer. EN omfanget splitting (energiforskjell) avhenger av arten av kation og ligander.
- 3. Befolkningen i det delte undernivået med elektroner adlyder tre generelle prinsipper - Pauli-forbudet, jakten på en minimum orbital energi og Hunds regel - jakten på maksimalt totalt spinn. Men i mange tilfeller kommer de to siste prinsippene i konflikt, og resultatet avhenger av forholdet mellom splittingsparameteren og paringsenergien. Paringsenergien P er energien som må brukes for å overvinne interelektronavstøtingen når et uparet elektron overføres til en orbital der det allerede er et annet elektron:
Hvis< P, то энергетически выгоднее заселение по правилу Хунда, даже если при этом приходится переводить электрон на более высокий подуровень. Если >P, så er det energimessig mer lønnsomt å befolke det lavere undernivået, selv om dette krever sammenkobling av elektroner. For noen kationer er to forskjellige tilstander mulige: en høyspinntilstand (HS) eller en tilstand i et svakt felt (< P), и низкоспиновое (НС), или состояние в сильном поле (>R). De er forskjellige i magnetiske egenskaper, ioniske radier og bindingsstyrke (se nedenfor). Høyspinntilstanden tilsvarer et større magnetisk moment, ionene i denne tilstanden er paramagnetiske (stoffet trekkes inn i magnetfeltet). I lavspinn-tilstanden avtar det magnetiske momentet, og hvis alle elektronene er sammenkoblet, så er det magnetiske momentet null, og stoffet er diamagnetisk (skjøvet ut av magnetfeltet).
4.2.3 Splitting av d-undernivået i ulike miljøer. For å forstå årsaken til splitting må man vurdere formen på elektronskyer i rommet, men figurene er ikke gitt her, de står i alle lærebøker. Tenk først på det oktaedriske miljøet, det enkleste og viktigste tilfellet. Seks identiske ligander er lokalisert rundt kationen langs tre gjensidig vinkelrette koordinatakser: venstre, høyre, foran, bak, topp, bunn. Deretter deles fem d-orbitaler i to grupper. d-orbitaler (d z 2 og d x 2 - y 2) forlenges direkte til liganden, frastøter dem, og deres energi øker, og d-orbitaler (d xy, d xz, d yz) forlenges forbi liganden - langs halveringslinjene til koordinatvinklene frastøter lite, og energien deres avtar. For å holde gjennomsnittsenergien uendret, går tre d-orbitaler ned med 2/5 og to d-orbitaler går opp med 3/5.
I et tetraedrisk miljø, tvert imot, har d-orbitaler økt energi, og d-orbitaler har en lavere. Dette kan forstås hvis vi kombinerer tetraederet med oktaederet slik at deres symmetriakser faller sammen. Da er toppene til tetraederet i stedet for oktaederets flater, dvs. fjernet fra aksene som d-orbitalene strekker seg langs.
På ris. 3 splittingsskjemaer er gitt for flere av de enkleste høysymmetriske koordinasjonene med de samme ligander. Hvis symmetrien er lav (for eksempel hvis alle ligander er forskjellige), vil alle fem d-orbitaler ha forskjellige energier. For en firkantet bipyramide (strukket oktaeder) og en firkantet pyramide plottes ikke nivåer, fordi deres posisjon avhenger av strekningen av oktaederet. Det begrensende tilfellet med å strekke oktaederet er en flat firkant. Ved å koble orbitalene med samme navn til oktaedriske og kvadratiske komplekser med rette linjer, får vi eventuelle mellomstadier av transformasjonen av oktaederet til en firkant når to toppunkter fjernes.
Figur 3. Splitting av d-subnivået i et felt med forskjellig symmetri
4.2.4 Faktorer som påvirker mengden av splitting. TKP tillater ikke å beregne verdiene og P, men tillater å finne fra den eksperimentelle målingen ...
Det er umulig å måle EXP direkte, men det kan estimeres ved å sammenligne termokjemiske data for forskjellige stoffer. For eksempel endrer hydreringsvarmen til dobbeltladede kationer av 3d-elementer seg over perioden i sikksakk med maksima ved V 2+ og Ni 2+. Sammenligning med ESCR-tabellen (se over) viser at dette stemmer godt overens med endringen i ESCR i et svakt oktaedrisk felt. Derav konklusjonen: dobbeltladede kationer av d-elementer fra den fjerde perioden danner, med overflødig vann, høyspinnkomplekser med CN 6 av den oktaedriske formen. Når man kjenner spinntilstanden, kan man beregne det magnetiske momentet til ionet, velge riktig verdi av den ioniske radiusen og forutsi metall-ligand-avstandene. Ved å måle avviket til den termiske effekten fra den glatte Ca 2+ - Mn 2+ - Zn 2+ linjen og ta den som ESCP, kan vi finne splittingsparameteren, og bruke den til å forutsi absorpsjonsspekteret. Omvendt kan absorpsjonsspekteret brukes til å bestemme koordinasjonen, spinntilstanden, ESCR og så videre. Dermed etablerte TCP forholdet mellom slike heterogene og tilsynelatende urelaterte egenskaper som de termiske effektene av reaksjoner, geometrien til strukturen, optiske og magnetiske egenskaper, uten å bruke komplekse kvantekjemiske beregninger.
4.2.7 Foretrukne koordinasjoner av d-element kationer (tabell 9). Bare for tre elektroniske konfigurasjoner (d 0 , BC d 5 og d 10) er ESCP lik null i ethvert miljø, og for de fleste konfigurasjoner er stabiliseringsenergien i et oktaedrisk felt større enn i et tetraedrisk. La oss beregne forskjellen deres (preferanseenergi for oktaedrisk koordinasjon) ved å bruke konfigurasjonen d 3 (eller d 8) som et eksempel: 6 o /5 - 4 t /5 = 6 o /5 - (4/9)*(4/ 5) o = 38 o /45 = 0,84o. Denne verdien, i størrelsesorden hundrevis av kJ/mol, er et meget alvorlig bidrag til bindingsenergien. Derfor, for ioner med d 3-konfigurasjonen, er oktaedrisk koordinasjon spesielt karakteristisk, mens tetraedrisk koordinasjon nesten aldri blir funnet. Oktaedrisk koordinasjon er også karakteristisk for de fleste andre elektroniske konfigurasjoner, spesielt siden den vanligvis tilsvarer maksimalt mulig CN, tatt i betraktning forholdet mellom kation- og ligandradius. For konfigurasjoner d 0 , BC d 5 og d 10 predikerer ikke TAP en preferanse for noen koordinering, så tetraedrisk koordinering er noe mer vanlig hos dem, men likevel er det ikke å foretrekke (bortsett fra i tilfeller hvor høyere CN er umulig pga. størrelsesbegrensninger eller i henhold til betingelsen om koordinasjonsbalanse). Høye CN-er (7-12) på grunn av geometriske hindringer finnes sjelden i kationer av d-elementer: bare i de største av dem med små ligander. Her blir de nesten ikke vurdert, pga. deres analyse er vanskeligere, og resultatene er mindre informative. I disse tilfellene er enkle "ioniske" representasjoner vanligvis tilstrekkelig til å forutsi koordinering.
TCH tillater imidlertid forutse utseendet i visse tilfeller koordinasjoner som motsier den ioniske modellen, det vil si ikke gir maksimale vinkler for en gitt CF: firkantet pyramide, firkantet, trekantet prisme. Disse koordinasjonene oppstår på grunn av dirigerte kovalente bindinger, men på språket til TCP er dette beskrevet gjennom ESCR.
Tenk på et kation med fire d-elektroner i et svakt oktaedrisk felt. Den forventer konfigurasjonen d 3 d 1 . Dermed er en av de to d-orbitalene okkupert. Men disse to orbitaler samhandler med forskjellige ligander: z 2 -orbital frastøter mest av alle to ligander som ligger langs z-aksen, og x 2 -y 2 -orbital, tvert imot, frastøter fire ligander i xoy-planet. Følgelig er liganden delt inn i to grupper, forskjellig i frastøtningsstyrken, og dermed i lengden på bindingen, og oktaeder kan ikke være riktig. Denne konklusjonen er et spesielt tilfelle av kvantemekanikk Jahn-Teller teoremer, som i en forenklet formulering lyder: hvis orbitalene til ett undernivå er ujevnt befolket i en ikke-lineær struktur, er en slik struktur ustabil og forvrengt slik at undernivået deler seg. Det er ingen instruksjoner her hva er det må være en forvrengning, men i et spesielt tilfelle er det ikke vanskelig å forutse. Av de to måtene å forvrenge oktaederet: kompresjon (2 korte bindinger + 4 lange bindinger) og strekking (4 korte bindinger + 2 lange bindinger), er den andre å foretrekke, fordi gir mer CC. Han er den vanligste. Strekkingen av oktaederet langs z-aksen kan føre til følgende varianter av koordinering: kvadratisk dipyramide (cn 4+2 eller 4+1+1); kvadratisk pyramide (CC 4+1); flat firkant (CH 4). Forvrengning resulterer i ytterligere deling av d-undernivået (se fig. ris. 3): x 2 -y 2 orbitalen frastøter spesielt sterkt de gjenværende liganden og øker energien, men z 2 orbitalen reduserer energien kraftig, fordi de ligandene som interagerte med den forlot. Elektronet opptar et lavere undernivå, og stabiliseringsenergien til krystallfeltet kompenserer for svekkelsen av ionbindingen på grunn av fjerning av to ligander.
Vil Jahn-Teller-effekten bli observert hvis orbitalene er ujevnt befolket Nedre undernivå (for eksempel med konfigurasjoner d 1 eller d 2 i et oktaedrisk felt)? Teoretisk - ja. I praksis samhandler imidlertid disse orbitalene svakt med ligander og deler seg svakt; derfor er forvrengningen liten og ofte maskert av termiske vibrasjoner. Det samme gjelder forvrengninger av tetraedriske strukturer. For å undertrykke vibrasjoner kan du avkjøle stoffet. Men så krystalliserer det seg, og det kan også oppstå forvrengninger i krystallen av andre årsaker – på grunn av forholdene med pakking med naboioner eller molekyler. Derfor Jahn-Teller-effekten er viktigst hvor d-orbitaler er ujevnt befolket i et oktaedrisk miljø, det vil si totalt for tre konfigurasjoner: BC d 4 , HC d 7 og d 9. Ioner med slike konfigurasjoner blir ofte referert til som Jahn-Teller-ioner. Som en øvelse kan du liste opp slike ioner (i oksidasjonstilstand 2+ og 3+) og bestemme i hvilke elektroniske konfigurasjoner Jahn-Teller-effekten vil være spesielt merkbar i tetraedrisk koordinasjon.
For d 8-konfigurasjonen i et svakt oktaedrisk felt er en slik forvrengning ugunstig, siden Av de to elektronene som befolker d-subnivået, er det bare en som senker energien, mens den andre hever den. Men med sterk splittelse (i et sterkt ligandfelt og når to naboer er fullstendig fjernet), kan elektroner pare seg på et lavere z 2-subnivå, og da blir kvadratisk koordinasjon gunstig for d 8-konfigurasjonen.
Fra delingsskjemaene som er avbildet i ris. 3, er den største reduksjonen i energien til det nedre undernivået observert i et trekantet prisme. Men gevinsten i ESC sammenlignet med oktaedrisk koordinering er liten, men tapet på grunn av tilnærmingen til like ioner er betydelig. Derfor er trekant-prismatisk koordinasjon sjelden. For at den skal bli mer stabil enn den oktaedriske, kreves følgende forhold samtidig:
Tabell 9. Koordinasjonspreferanser for kationer av d-elementer (hvis størrelse og betingelser for koordinasjonsbalanse, kompatibilitet av koordinasjonsgrupper tillater det). Fet skrift indikerer tilfeller der denne koordineringen er spesielt foretrukket. Store kationer kan ha CN>6, men dette er ikke tatt hensyn til i tabellen.
Elektronisk
Foretrukket koordinering
d1, d2, d3, NS d 4, NS d 5,NS d 6, BC d 6 , Søn d 7, Søn d 8
BC d4, NC d7, d9
Strukket firkantet (di) pyramide, noen ganger kvadratisk: CC 4+2, 4+1(+1), 4
5d 2 , 4d 2 , noen ganger 5d 1 , 4d 1
Trekantet prisme (med lav elektronegativitet av ligander), firkantet antiprisme
Enhver svært symmetrisk koordinasjon: oktaeder, tetraeder...
Det samme, men på grunn av den økte elektronegativiteten til slike ioner (lav ionisitet av bindingen) og det lille antallet frie AO-er, er det en preferanse for lave CN-er, litt høyere enn oksidasjonstilstanden: 4, 3, 2.
Det samme som for HS d 5 , men med økt EC av kationen med ligander av -donortypen, er forvrengninger av oktaeder karakteristiske, spesielt CN 1+4(+1), 2+2+2, 3+3, med korte bindinger i cis-posisjon (cm. klausul 4.2)
- - en veldig stor splittende verdi (derfor observeres slik koordinering bare i 4d- og 5d-, men ikke 3d-elementer);
- - tilstedeværelsen av et lite antall elektroner på d-undernivået, fortrinnsvis to (med et annet antall elektroner er det ingen signifikant gevinst i ESCP sammenlignet med oktaederet);
- - små effektive ladninger av liganden og store bindingslengder (for å redusere gjensidig frastøting av nære ligander).
Typiske eksempler hvor alle disse betingelsene er oppfylt er molybdensulfid (4+) 2 og dets ditiolatkomplekser, for eksempel 2-. Hvis vi erstatter molybden med et element fra 4. periode, eller svovel med oksygen, eller endrer oksidasjonstilstanden til molybden, oppnås ikke trekantede prismer. I samme oksidasjonstilstand med små ligander som skaper et sterkt felt, har molybden noen ganger også en kvadratisk antiprismekoordinasjon: 4- , men denne formen er også forutsagt av den ioniske modellen uten å ta hensyn til den elektroniske strukturen.
De siste årene har det dukket opp pålitelige data om eksistensen av trekantede prismatiske strukturer selv ved den høyeste oksidasjonstilstanden til d-elementet, for eksempel 2-. Dette er uforklarlig innenfor rammen av TSP og AIM. En forklaring er gitt av teorien om molekylære orbitaler, men den ligger utenfor dette kursets omfang.
- 4.2 Koordinasjonskjemi av fullvalente kationer av d-elementer
- 4.2.1 Generelle egenskaper. I høyeste grad av oksidasjon har kationer av d-elementer et 8-elektrons skall av en inert gass, dvs. er sfærisk symmetriske, og det ser ut til at de ikke burde ha noen funksjoner i koordinasjonskjemien - et svært symmetrisk miljø forventes og CN er maksimalt mulig avhengig av størrelsen. Faktisk, for de minste av dem, som Cr(6+) og Mn(7+), er tetraedrisk koordinering eksepsjonelt karakteristisk, for de største med små ligander (zirkoniumfluorokomplekser) - CN 7-9, og for de fleste - oktaedrisk koordinering . Men tilstedeværelsen av ni tomme AO-er på det ytre nivået gjør dem i det oktaedriske komplekset ikke bare -akseptorer (på grunn av s, p og to d-orbitaler), men også -akseptorer (på grunn av tre d-orbitaler), og dette fører til funksjonene som er omtalt nedenfor.
- 4.2.2 Asymmetri -binding i oktaedriske komplekser. I det oktaedriske komplekset til d 0 -kationen med ligander av -donortypen, konkurrerer 6 (eller til og med 12) elektronpar med ligander om tre tomme d-orbitaler. Alle av dem, i henhold til Pauli-prinsippet, kan ikke overføres til disse tre orbitalene. Av disse 12 orbitalene kan tre symmetriske "gruppeorbitaler" kombineres. Men det er vanligvis energimessig mer lønnsomt å velge en, to eller tre av de 6 naboene og knytte korte sterke +-bånd med dem.
Ris. 4.
Fra ris. 4 det kan sees at overlappingen av orbitaler med en av naboene og overføring av elektrontetthet fra dette oksygenatomet til d-orbitalen forhindrer, ifølge Pauli-prinsippet, kommunikasjon med den motsatte naboen. Jo sterkere og kortere forbindelsen er med en av naboene, desto svakere og lengre er forbindelsen med den motsatte (trans-) partneren. Derfor bør korte sterke bindinger være plassert side ved side (i cis-posisjoner i forhold til hverandre), og lange svake bør være motsatt. Merk at tilnærmingen til atomer på grunn av dannelsen en-forbindelser letter utdanning den andre - lenker med det samme nabo, i et vinkelrett plan. Dette er ikke nødvendigvis en trippelbinding (+to), rekkefølgen på hver av de tre bindingene kan være mindre enn én, men i alle fall er den totale bindingsrekkefølgen ganske stor.
Dette fører til en forskyvning av kationen fra midten av oktaederet til ett, to eller tre hjørner, og dets CN er henholdsvis 1 + 4 (+1), 2 + 2 + 2 eller 3+3. Med en liten forskyvning utvendig konturene til oktaederet kan forbli nesten regelmessige, men med en sterk forskyvning går det sjette toppunktet tapt, og en firkantet pyramide oppnås.
Betingelser for forekomsten av en slik forvrengning av oktaedre:
- - ligand - donor (oksygen, fluor, nitridion, men ikke ammoniakk og ikke amin);
- - metall - -akseptor (elektronisk konfigurasjon d 0 eller d 1, det vil si den høyeste oksidasjonstilstanden eller 1 mindre);
- - betydelig kovalens av bindingen, det vil si ikke en veldig stor forskjell i elektronegativitet, derfor, for oksygenforbindelser, er dette mer karakteristisk enn for fluorider, og for vanadium, molybden - mer karakteristisk enn for wolfram, niob, titan og til og med mer så tantal, zirkonium, hafnium.
- 4.2.3 Effekt av -binding i oktaedre på anionkoordinasjon og strukturforbindelse
Eksistensen av Vanadil og lignende grupper
Vanadium (+4), som har ett elektron på d-subnivået og derfor har to tomme d-orbitaler, kan delta i to -bindinger. Det er mest fordelaktig å danne begge -bindinger med det samme et oksygenatom. Det viser seg en veldig sterk gruppering O \u003d V 2+ - vanadyl, hvor bindingsrekkefølgen er praktisk talt lik 2, i stand til å gå uendret fra ett stoff til et annet. I en sur vandig løsning er det ikke protonert, fordi oksygenatomet er valensmettet. Typiske former for vanadium (+4) i sure løsninger og krystallinske hydrater er 2+ og 2+. I hydroksydet, i stedet for vannmolekyler, er det brohydroksylgrupper: 1, og i en alkalisk løsning, sannsynligvis 2-.
Asymmetri -binding og sammenheng av strukturer
Oksygenanionet som deltar i en dobbeltbinding med d 0 -kationen er valensmettet og trenger ikke kombineres med det andre kationen, det vil si å være brodannende. På den brytes strukturen av og tilkoblingen reduseres. For eksempel, i V 2 O 5 har ett av oksygenatomene avstander til vanadiumatomer på 1,56 A (veldig sterk, nesten dobbeltbinding) og 2,84 A - nesten fullstendig fravær av en binding. Derfor er vanadiumoksid (5) lagdelt. Derav dets relativt lave smeltepunkt og høye reaktivitet sammenlignet med niobiumoksid (5): for eksempel V 2 O 5 Lettløselig i både syrer og alkalier. Hvis man ikke kjenner til særegenhetene ved koordineringskjemien til vanadium og molybden, er det vanskelig å forutse disse resultatene. Forutsatt en CN av kationer på 5 eller 6, ville man forvente at alle anioner har en CN på 2 (eller enda mer), strukturen er tverrbundet i tre dimensjoner, stoffet er ildfast, ikke-flyktig og inaktivt. Faktisk er 2 bare gjennomsnitt CN av oksygen, mens de virkelige er 1, 2 og 3, og derfor er tilkoblingen senket.
4.2.4 Asymmetri-binding og spesielle elektriske og optiske egenskaper. Hvis det er uendelige lineære kjeder i strukturen -О-М-О-М-О-М- (MO 6 oktaedre er forbundet med toppunkter), der M er en d 0 -kation utsatt for -bindingsasymmetri, så er forskyvningen av en av kationene til en av naboene (for eksempel til venstre) vil føre til, for å opprettholde den lokale valensbalansen, forskyvning av nabokationer i samme retning. Korte og lange bindinger vil veksle i kjeden, tyngdepunktet til positive ladninger vil ikke lenger falle sammen med tyngdepunktet til negative, strukturen vil bli polar (pluss til venstre, minus til høyre). Hvis en slik forskyvning er liten, kan pålegging av et eksternt elektrisk felt med samme polaritet føre til at ionene skifter i motsatt retning. Deretter, hvis feltet fjernes, vil den omvendte skjevheten ikke oppstå, siden skiftet av kationer til høyre er ikke verre enn skiftet til venstre. Et slikt fenomen - eksistensen av sin egen polaritet, hvis retning kan reorienteres av et eksternt elektrisk felt - kalles ferroelektrisitet, og selve materialet ferroelektrisk. Indre polarisering eksisterer vanligvis bare ved lave temperaturer, og oppvarming fremmer en overgang til en mer symmetrisk ikke-polar struktur uten forskyvninger (eller med erstatning av konstante forskyvninger med intense termiske vibrasjoner). Ferroelektrikk har mange praktiske anvendelser basert på konvertering av elektriske signaler til mekaniske og omvendt (mikrofoner, telefoner, hydrofoner, ultralydsendere, trykk- og akselerasjonssensorer), elektro-optiske effekter (kontroll av en laserstråle ved hjelp av et elektrisk felt), minneeffekt , etc. For at forskyvninger skal eksistere, men være små, trenge noen, ikke for stor og ikke for liten, grad av bindingsionisitet. Perfekt passform for dette titanater og niobater. I oksygenforbindelser av vanadium og molybden er kationforskyvninger og oktaederforvrengninger vanligvis så store (se ovenfor) at de ikke kan reverseres av et ytre felt (ett toppunkt av et oktaeder går ofte helt tapt, og man får en firkantet pyramide). Tvert imot, når oksygen erstattes med fluor, eller niob med tantal, eller titan med zirkonium, øker ionisiteten til bindingen, kovalente effekter svekkes, og arrangementet av ioner blir upolært selv ved lave temperaturer. Delvis substitusjon (dannelse av faste løsninger) lar deg jevnt kontrollere egenskapene. De viktigste ferroelektriske: BaTiO 3 , Pb(Ti 1-x Zr x)O 3 , KNbO 3 , LiNbO 3 , PbNb 2 O 6 .
Prosjektoppgave
Som instruert av læreren, finn strukturell informasjon om et bestemt stoff på Internett, last ned en publikasjon (eller enda bedre, en cif-krystallografisk informasjonsfil), analyser strukturen ved å bruke Diamond-programmet og gi dens skriftlige beskrivelse i henhold til følgende skjema: formel, krystallografiske data, forskningsmetode (enkeltkrystall eller pulver, røntgendiffraksjon eller nøytrondiffraksjon), koordinering av hver komponent (ikke bare CN, men en detaljert karakteristikk, som tar hensyn til ikke-ekvivalens av bindinger, bindingsvinkler, brøkdeler populasjoner, hvis noen), beregning av bindingsvalenser og kontroll av den lokale valensbalansen, karakterisering av tilkobling og hvordan den implementeres (forbindelse av polyedre med toppunkter, kanter eller flater), personlig vurdering av påliteligheten til dataene, grad av konsistens med prinsippene angitt i denne håndboken, bibliografisk referanse. Anbefalinger for arbeid med lignende materialer er i manualene.
Søkeanbefalinger. Spesialiserte vitenskapelige søkemotorer www.scopus.com og scholar.google.com. Den første er kun tilgjengelig via SFedU-proxyserveren og gir gratis tilgang til de fullstendige tekstene til mange tidsskrifter publisert av Elsevier. I tillegg er fulltekster fra tidsskriftene til American Chemical Society (pubs.acs.org) og Physical Society, London Royal Society of Chemistry (www.rsc.org) tilgjengelige via SFedU-proxyserveren. Tidsskriftene til International Union of Crystallographers (www.iucr.org) gir ikke gratis tilgang til de fleste artikler, men cif er alltid tilgjengelig. Å åpne cif Diamond er uforlignelig enklere enn å legge inn de digitale dataene fra artikkelen fra tastaturet.En demoversjon av Diamond er tilgjengelig på avdelingen.
Midterm kontrolltest nr. 4
Testen inneholder 6 oppgaver, som tar 3 minutter å gjennomføre. Velg det mest riktige, etter din mening, svaralternativet og merk det med et hvilket som helst ikon i svararket.
1. Hvilket par av analoge elementer har størst forskjell i radier?
2. Ioniske radier av kationer av d-elementer i samme oksidasjonstilstand over perioden med økende serienummer
avta monotont
øke monotont
har lokale ytterpunkter med en generell nedadgående trend
har lokale ytterpunkter med en generell oppadgående trend
3. Parameteren for splitting av krystallfeltet øker
etter undergruppe fra topp til bunn
under overgangen fra ligander - -akseptorer til ligander - -donorer
med økende kationradius
med en nedgang i HF
4. Hvis du trenger å spesifisere bare én viktigste faktor som bestemmer forskjellene i koordineringen av Mn(2+) og Mn(7+) (selv om alt er viktig!), er det
antall gratis JSC-er
ionisk radius
magnetisk moment
5. Hvis du trenger å spesifisere kun én viktigste faktor som bestemmer forskjellene i koordineringen av Mn(2+), Mn(3+) og Mn(4+) (selv om alt er viktig!), er det
antall gratis JSC-er
krystallstabiliseringsenergi. felt
ionisk radius
magnetisk moment
6. Forskyvning fra sentrum av oktaederet (opp til dets transformasjon til en firkantet pyramide) er mest karakteristisk for
Svarskjema
Vurderingskriterium: Hver oppgave er verdt 5 poeng. Prøven regnes som bestått dersom du får 25 poeng.