Em reações redox, substâncias orgânicas mais freqüentemente exibem as propriedades de agentes redutores, enquanto eles próprios são oxidados. A facilidade de oxidação de compostos orgânicos depende da disponibilidade de elétrons ao interagir com um agente oxidante. Todos os fatores conhecidos que causam um aumento na densidade de elétrons nas moléculas de compostos orgânicos (por exemplo, efeitos indutivos positivos e mesoméricos) aumentarão sua capacidade de oxidação e vice-versa.
A tendência dos compostos orgânicos para oxidar aumenta com o crescimento de sua nucleofilicidade, que corresponde às seguintes linhas:
O crescimento da nucleofilicidade na série
Considerar reações redox representantes das classes mais importantes matéria orgânica com alguns agentes oxidantes inorgânicos.
oxidação de alceno
Com oxidação suave, os alcenos são convertidos em glicóis (álcoois diídricos). Os átomos redutores nessas reações são átomos de carbono ligados por uma ligação dupla.
A reação com uma solução de permanganato de potássio ocorre em meio neutro ou ligeiramente alcalino da seguinte forma:
3C 2 H 4 + 2KMnO 4 + 4H 2 O → 3CH 2 OH–CH 2 OH + 2MnO 2 + 2KOH
Em condições mais severas, a oxidação leva à quebra da cadeia carbônica na ligação dupla e à formação de dois ácidos (em meio fortemente alcalino, dois sais) ou um ácido e dióxido de carbono (em meio fortemente alcalino, um sal e um carbonato):
1) 5CH 3 CH=CHCH 2 CH 3 + 8KMnO 4 + 12H 2 SO 4 → 5CH 3 COOH + 5C 2 H 5 COOH + 8MnSO 4 + 4K 2 SO 4 + 17H 2 O
2) 5CH 3 CH=CH 2 + 10KMnO 4 + 15H 2 SO 4 → 5CH 3 COOH + 5CO 2 + 10MnSO 4 + 5K 2 SO 4 + 20H 2 O
3) CH 3 CH=CHCH 2 CH 3 + 8KMnO 4 + 10KOH → CH 3 COOK + C 2 H 5 COOK + 6H 2 O + 8K 2 MnO 4
4) CH 3 CH \u003d CH 2 + 10KMnO 4 + 13KOH → CH 3 COOK + K 2 CO 3 + 8H 2 O + 10K 2 MnO 4
O dicromato de potássio em meio ácido sulfúrico oxida os alcenos de forma semelhante às reações 1 e 2.
Durante a oxidação de alcenos, nos quais os átomos de carbono na ligação dupla contêm dois radicais de carbono, duas cetonas são formadas:


Oxidação de alcino
Os alcinos oxidam sob condições um pouco mais severas do que os alcenos, então eles geralmente oxidam com a ligação tripla quebrando a cadeia de carbono. Como no caso dos alcenos, os átomos redutores aqui são átomos de carbono ligados por uma ligação múltipla. Como resultado das reações, ácidos e dióxido de carbono são formados. A oxidação pode ser realizada com permanganato ou dicromato de potássio em meio ácido, por exemplo:
5CH 3 C≡CH + 8KMnO 4 + 12H 2 SO 4 → 5CH 3 COOH + 5CO 2 + 8MnSO 4 + 4K 2 SO 4 + 12H 2 O

O acetileno pode ser oxidado com permanganato de potássio em meio neutro a oxalato de potássio:
3CH≡CH +8KMnO 4 → 3KOOC –COOK +8MnO 2 +2KOH +2H 2 O
Em um ambiente ácido, a oxidação vai para o ácido oxálico ou dióxido de carbono:
5CH≡CH + 8KMnO 4 + 12H 2 SO 4 → 5HOOC -COOH + 8MnSO 4 + 4K 2 SO 4 + 12H 2 O
CH≡CH + 2KMnO 4 + 3H 2 SO 4 → 2CO 2 + 2MnSO 4 + 4H 2 O + K 2 SO 4
Oxidação de homólogos de benzeno
O benzeno não oxida mesmo em condições bastante severas. Homólogos de benzeno podem ser oxidados com uma solução de permanganato de potássio em meio neutro a benzoato de potássio:
C 6 H 5 CH 3 + 2KMnO 4 → C 6 H 5 COOK + 2MnO 2 + KOH + H 2 O
C 6 H 5 CH 2 CH 3 + 4KMnO 4 → C 6 H 5 COOK + K 2 CO 3 + 2H 2 O + 4MnO 2 + KOH
A oxidação de homólogos de benzeno com dicromato ou permanganato de potássio em meio ácido leva à formação de ácido benzóico.
5C 6 H 5 CH 3 + 6KMnO 4 +9 H 2 SO 4 → 5C 6 H 5 COOH + 6MnSO 4 + 3K 2 SO 4 + 14H 2 O
5C 6 H 5 –C 2 H 5 + 12KMnO 4 + 18H 2 SO 4 → 5C 6 H 5 COOH + 5CO 2 + 12MnSO 4 + 6K 2 SO 4 + 28H 2 O



Oxidação do álcool
Os produtos diretos da oxidação dos álcoois primários são os aldeídos, enquanto os dos álcoois secundários são as cetonas.
Os aldeídos formados durante a oxidação de álcoois são facilmente oxidados a ácidos; portanto, aldeídos de álcoois primários são obtidos por oxidação com dicromato de potássio em meio ácido no ponto de ebulição do aldeído. Evaporando, os aldeídos não têm tempo para oxidar.
3C 2 H 5 OH + K 2 Cr 2 O 7 + 4H 2 SO 4 → 3CH 3 CHO + K 2 SO 4 + Cr 2 (SO 4) 3 + 7H 2 O
Com excesso de agente oxidante (KMnO 4, K 2 Cr 2 O 7) em qualquer meio, os álcoois primários são oxidados a ácidos carboxílicos ou seus sais, e os álcoois secundários a cetonas.
5C 2 H 5 OH + 4KMnO 4 + 6H 2 SO 4 → 5CH 3 COOH + 4MnSO 4 + 2K 2 SO 4 + 11H 2 O
3CH 3 -CH 2 OH + 2K 2 Cr 2 O 7 + 8H 2 SO 4 → 3CH 3 -COOH + 2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O
Os álcoois terciários não são oxidados nessas condições, mas o álcool metílico é oxidado a dióxido de carbono.

O álcool diídrico, etileno glicol HOCH 2 -CH 2 OH, quando aquecido em meio ácido com uma solução de KMnO 4 ou K 2 Cr 2 O 7, é facilmente oxidado a ácido oxálico e em meio neutro a oxalato de potássio.
5CH 2 (OH) - CH 2 (OH) + 8KMnO 4 + 12H 2 SO 4 → 5HOOC -COOH + 8MnSO 4 + 4K 2 SO 4 + 22H 2 O
3CH 2 (OH) - CH 2 (OH) + 8KMnO 4 → 3KOOC -COOK + 8MnO 2 + 2KOH + 8H 2 O
Oxidação de aldeídos e cetonas
Os aldeídos são agentes redutores bastante fortes e, portanto, são facilmente oxidados por vários agentes oxidantes, por exemplo: KMnO 4, K 2 Cr 2 O 7, OH, Cu (OH) 2. Todas as reações ocorrem quando aquecidas:
3CH 3 CHO + 2KMnO 4 → CH 3 COOH + 2CH 3 COOK + 2MnO 2 + H 2 O
3CH 3 CHO + K 2 Cr 2 O 7 + 4H 2 SO 4 → 3CH 3 COOH + Cr 2 (SO 4) 3 + 7H 2 O
CH 3 CHO + 2KMnO 4 + 3KOH → CH 3 COOK + 2K 2 MnO 4 + 2H 2 O
5CH 3 CHO + 2KMnO 4 + 3H 2 SO 4 → 5CH 3 COOH + 2MnSO 4 + K 2 SO 4 + 3H 2 O
CH 3 CHO + Br 2 + 3NaOH → CH 3 COONa + 2NaBr + 2H 2 O

reação do espelho de prata
Com uma solução de amônia de óxido de prata, os aldeídos são oxidados a ácidos carboxílicos, que dão sais de amônio em uma solução de amônia (reação de “espelho de prata”):
CH 3 CH \u003d O + 2OH → CH 3 COONH 4 + 2Ag + H 2 O + 3NH 3
CH 3 -CH \u003d O + 2Cu (OH) 2 → CH 3 COOH + Cu 2 O + 2H 2 O
O aldeído fórmico (formaldeído) é oxidado, por via de regra, ao dióxido de carbono:
5HCOH + 4KMnO4 (cabana) + 6H 2 SO 4 → 4MnSO 4 + 2K 2 SO 4 + 5CO 2 + 11H 2 O
3CH 2 O + 2K 2 Cr 2 O 7 + 8H 2 SO 4 → 3CO 2 + 2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O
HCHO + 4OH → (NH 4) 2 CO 3 + 4Ag↓ + 2H 2 O + 6NH 3
HCOH + 4Cu(OH) 2 → CO 2 + 2Cu 2 O↓+ 5H 2 O
As cetonas são oxidadas sob condições severas por agentes oxidantes fortes com a quebra das ligações C-C e dão origem a misturas de ácidos:

ácidos carboxílicos. Entre os ácidos, os ácidos fórmico e oxálico têm fortes propriedades redutoras, que são oxidados a dióxido de carbono.
HCOOH + HgCl 2 \u003d CO 2 + Hg + 2HCl
HCOOH + Cl 2 \u003d CO 2 + 2HCl
HOOC-COOH + Cl 2 \u003d 2CO 2 + 2HCl
Ácido fórmico, além das propriedades ácidas, também exibe algumas propriedades dos aldeídos, em particular, redutoras. Em seguida, é oxidado a dióxido de carbono. Por exemplo:
2KMnO4 + 5HCOOH + 3H2SO4 → K2SO4 + 2MnSO4 + 5CO2 + 8H2O
Quando aquecido com agentes desidratantes fortes (H2SO4 (conc.) ou P4O10) decompõe-se:
HCOOH →(t)CO + H2O
Oxidação catalítica de alcanos:

Oxidação catalítica de alcenos:

Oxidação do fenol:


Alcinos com uma ligação tripla não terminal servem como uma fonte potencial para a síntese de 1,2-dicetonas sob a ação de um agente oxidante adequado. No entanto, ainda não foi encontrado nenhum reagente universal que cause a oxidação de uma ligação tripla carbono-carbono a um grupo 1,2-dicarbonila. RuO 4 proposto para este fim, óxido de rutênio (VIII), é muito caro e muitas vezes causa degradação oxidativa adicional de 1,2-dicetonas em ácidos carboxílicos. Quando os acetilenos dissubstituídos reagem com agentes oxidantes fortes como o permanganato de potássio, apenas em um meio completamente neutro a pH 7–8 a 0 С a oxidação pode ser interrompida no estágio de formação de -dicetona. Assim, por exemplo, o ácido estearólico a pH 7,5 é oxidado a α-dicetona. Na maioria dos casos, a oxidação é acompanhada pela quebra da ligação tripla com a formação de ácidos carboxílicos:

O rendimento dos produtos da degradação oxidativa dos alcinos é baixo e essa reação não desempenha um papel significativo na síntese orgânica. É usado apenas para provar a estrutura do ácido acetilênico natural contido nas folhas de plantas tropicais da América Central. Durante sua destruição oxidativa, dois ácidos foram isolados - láurico e adípico. Isso significa que o ácido original é o ácido 6-octadécico com um esqueleto de carbono normal de dezessete carbonos:
Muito mais importante é o acoplamento oxidativo de alcinos-1 catalisado por sais de cobre (a reação de Glaser-Eglinton). Em 1870, Glaser descobriu que uma suspensão de cobre (I) acetilenito em álcool é oxidada pelo oxigênio atmosférico para formar 1,3-diinos:

Para a oxidação de acetilenetos de cobre (I), o hexacianoferrato de potássio (III) K 3 em DME ou DMF é mais eficaz como agente oxidante. Em 1959, Eglinton propôs uma modificação muito mais conveniente da condensação oxidativa dos alcinos. Alcino é oxidado com acetato de cobre(II) em uma solução de piridina a 60–70°C. A modificação de Eglinton provou ser extremamente útil para a síntese de poliinos macrocíclicos a partir de ,-diinos. Como ilustração, apresentamos a síntese de duas ciclopoliinas durante a condensação oxidativa da hexadiína-1,5 (F. Sondheimer, 1960):

Uma das poliínas é um produto da ciclotrimerização, a outra é um produto da ciclotetramerização da gesadiina-1,5 inicial. O trímero serve como reagente inicial para a síntese de -anulenos aromáticos (para mais detalhes sobre anéis, veja o Capítulo 12). Da mesma forma, nas mesmas condições da nonadiína-1,8, obtém-se seu dímero - 1,3,10,12-ciclooctadecatetraeno junto com trímero, tetrâmero e pentâmero:

Para obter diinos assimétricos, é usada a condensação de haloacetilenos com alcino-1 (alcino terminal) na presença de sais de cobre (I) e uma amina primária (acoplamento de acordo com Kadio–Chodkevich, 1957):

Os bromalquinos iniciais são obtidos pela ação do hipobromito de sódio sobre os alcinos-1 ou dos acetiletos de lítio e bromo:
O derivado de organocobre do alcino terminal é gerado diretamente na mistura de reação de Cu 2 Cl 2 e alcino-1.
6.3.4. Reações de adição eletrofílica de ligação tripla
As reações de adição eletrofílica à ligação tripla estão entre as reações mais típicas e importantes dos alcinos. Em contraste com a adição eletrofílica a alcenos, a aplicação sintética desse grande grupo de reações estava muito à frente do desenvolvimento de ideias teóricas sobre seu mecanismo. No entanto, nos últimos vinte anos, a situação mudou significativamente e, atualmente, é uma das áreas de rápido desenvolvimento da química orgânica física. O HOMO do alcino está localizado abaixo do HOMO do alceno (Seção 2), e esta circunstância na grande maioria dos casos predetermina a menor taxa de adição do agente eletrofílico ao alcino em comparação com o alceno. Outro fator que determina a diferença na reatividade de alcinos e alcenos em reações de adição eletrofílica é a estabilidade relativa dos intermediários que surgem quando uma espécie eletrofílica é adicionada a ligações triplas e duplas. Quando uma partícula eletrofílica H + ou E + é ligada a uma ligação dupla, um carbocátion cíclico ou aberto é formado (Cap. 5). A adição de H + ou E + a uma ligação tripla leva à formação de um cátion vinílico aberto ou cíclico. Em um cátion linear aberto de vinil, o átomo de carbono central está em sp-estado híbrido, enquanto vago R-orbital é ortogonal à ligação . Porque o sp-átomo de carbono híbrido do cátion vinil tem uma eletronegatividade mais alta em comparação com spÁtomo 2-híbrido do cátion alquil, o cátion vinil deve ser menos estável em comparação com o cátion alquil:

Os dados dos cálculos de mecânica quântica, bem como os dados termodinâmicos para a fase gasosa, obtidos por espectrometria de massa de alta pressão e espectroscopia de ressonância de ciclotron, estão de acordo com essas considerações. Na tabela. 6.3 mostra dados termodinâmicos para a formação de vários carbocátions e hidrocarbonetos, relacionados à fase gasosa a 25 С.
|
Carbocátion |
Δ H f ˚ kcal/mol |
|
| |
|
| |
|
|
A partir dos dados apresentados em Tal. 6.3, segue-se que o cátion vinil é 47 kcal/mol menos estável que o cátion etil contendo o mesmo número de átomos. A mesma conclusão pode ser tirada da entalpia de ionização na fase gasosa CH 3 CH 2 Cl e CH 2 =CHCl:
É fácil ver que a combinação de ambos os fatores - a maior energia do cátion vinil e o baixo HOMO do alcino - representa a menor reatividade dos alcinos em comparação com os alcenos nas reações de adição eletrofílica. Na tabela. 6.4 contém dados comparativos sobre a adição de halogênios, cloretos de sulfeno e selenila, ácido trifluoroacético e água a vários alcenos e alcinos que não contêm nenhum grupo funcional ativador ou desativador.
Tabela 6.4
Características comparativas de alcinos e alcenos
em reações de adição eletrofílica
|
substratos |
K alceno / K alcino |
|
|
Bromação em ácido acético |
CH 2 CH 2 /HCCH C 4 H 9 CH \u003d CH 2 / C 4 H 9 C CH C 6 H 5 CH \u003d CH 2 / C 6 H 5 C CH | |
|
Cloração em ácido acético |
C 6 H 5 CH \u003d CH 2 / C 6 H 5 C CH C 4 H 9 CH \u003d CH 2 / C 6 H 5 C CH C 2 H 5 C \u003d SNS 2 H 5 / C 2 H 5 C SS 2 H 5 | |
|
Adição de cloreto de 4-clorofenilsulfen P-ClС 6 H 4 SeCl |
CH 2 \u003d CH 2 / HC CH C 4 H 9 CH \u003d CH 2 / C 4 H 9 C CH C 6 H 5 CH \u003d CH 2 / C 6 H 5 C CH | |
|
Adição de cloreto de fenilseleno C 6 H 5 SeCl |
CH 2 \u003d CH 2 / HC CH C 4 H 9 CH \u003d CH 2 / C 4 H 9 C CH C 6 H 5 CH \u003d CH 2 / C 6 H 5 C CH | |
|
Adição de ácido trifluoroacético |
C 4 H 9 CH \u003d CH 2 / C 4 H 9 C CH C 6 H 5 CH \u003d CH 2 / C 6 H 5 C CH C 2 H 5 CH \u003d CH 2 / C 2 H 5 C CH | |
|
Hidratação catalisada por ácido |
C 4 H 9 CH \u003d CH 2 / C 4 H 9 C CH C 2 H 5 CH \u003d SNS 2 H 5 / C 2 H 5 C SS 2 H 5 C 6 H 5 CH \u003d CH 2 / C 6 H 5 C CH |
Segue-se desses dados que apenas a adição de agentes ácidos e água para ligações triplas e duplas ocorre em taxas semelhantes. A adição de halogênios, sulfencloretos e vários outros reagentes aos alcenos ocorre 10 2 - 10 5 vezes mais rápido do que aos alcinos. Isso significa que os hidrocarbonetos contendo ligações triplas e duplas não conjugadas adicionam seletivamente esses reagentes na ligação dupla, por exemplo:

Dados de hidratação comparativa de alcinos e alcenos devem ser tratados com cautela, pois a hidratação de alcinos requer catálise por íons mercúrio (II), que é ineficiente para a adição de água à ligação dupla. Portanto, os dados de hidratação das ligações tripla e dupla, a rigor, não são comparáveis.
A adição de halogênios, haletos de hidrogênio, cloretos de sulfen e outros agentes eletrofílicos pode ser realizada em etapas, que podem ser facilmente ilustradas usando os seguintes exemplos:



A tendência de oxidação dos compostos orgânicos está associada à presença de ligações múltiplas, grupos funcionais, átomos de hidrogênio no átomo de carbono que contém o grupo funcional. A oxidação sequencial de substâncias orgânicas pode ser representada como a seguinte cadeia de transformações:
Hidrocarboneto saturado → Hidrocarboneto insaturado → Álcool → Aldeído (cetona) → Ácido carboxílico →CO2 + H2O
A conexão genética entre classes de compostos orgânicos é apresentada aqui como uma série de reações redox que garantem a transição de uma classe de compostos orgânicos para outra. É completado pelos produtos da oxidação completa (combustão) de qualquer um dos representantes das classes de compostos orgânicos. A dependência da capacidade redox de uma substância orgânica em sua estrutura: A maior tendência dos compostos orgânicos à oxidação se deve à presença na molécula de substâncias: ligações múltiplas (é por isso que alcenos, alcinos, alcadienos são tão facilmente oxidados); certos grupos funcionais que podem ser facilmente oxidados (--SH, -OH (fenólico e álcool), -NH2; grupos alquil ativados localizados adjacentes a ligações múltiplas.
Por exemplo, o propeno pode ser oxidado ao aldeído insaturado acroleína com oxigênio atmosférico na presença de vapor de água em catalisadores de bismuto-molibdênio.
H2C═CH-CH3 → H2C═CH-COH
Bem como a oxidação do tolueno a ácido benzóico com permanganato de potássio em meio ácido. 5C6H5CH3 +6KMnO4 + 9H2SO4 → 5C6H5COOH + 3K2SO4 + 6MnSO4 +14H2O
a presença de átomos de hidrogênio no átomo de carbono contendo o grupo funcional. Um exemplo é a reatividade em reações de oxidação de álcoois primários, secundários e terciários por reatividade à oxidação.
Apesar de no decorrer de qualquer reação redox ocorrer oxidação e redução, as reações são classificadas dependendo do que acontece diretamente com o composto orgânico (se for oxidado, falam do processo de oxidação, se for reduzido, sobre o processo de redução).
Assim, na reação do etileno com o permanganato de potássio, o etileno será oxidado e o permanganato de potássio será reduzido. A reação é chamada de oxidação do etileno.
Ao estudar as características comparativas de compostos inorgânicos e orgânicos, nos familiarizamos com o uso do estado de oxidação (s.o.) (em química orgânica, principalmente carbono) e métodos para determiná-lo:
1) cálculo do s.d médio carbono na molécula da matéria orgânica: -8/3 +1 C3 H8 Esta abordagem é justificada se durante a reação todas as ligações químicas na matéria orgânica forem destruídas (combustão, decomposição completa).
2) definição de s.d. cada átomo de carbono:
Nesse caso, o estado de oxidação de qualquer átomo de carbono em um composto orgânico é igual à soma algébrica dos números de todas as ligações com átomos de elementos mais eletronegativos, levado em consideração o sinal “+” no átomo de carbono, e o número de ligações com átomos de hidrogênio (ou outro elemento mais eletropositivo), levado em consideração com o sinal "-" no átomo de carbono. Nesse caso, as ligações com átomos de carbono vizinhos não são levadas em consideração. Como exemplo mais simples, vamos determinar o estado de oxidação do carbono em uma molécula de metanol. O átomo de carbono está ligado a três átomos de hidrogênio (essas ligações são consideradas com o sinal "-"), uma ligação é com o átomo de oxigênio (é considerada com o sinal "+"). Obtemos: -3 + 1 = -2 Assim, o estado de oxidação do carbono no metanol é -2. O grau calculado de oxidação do carbono, embora seja um valor condicional, mas indica a natureza da mudança na densidade de elétrons na molécula, e sua mudança como resultado da reação indica um processo redox em andamento. Esclarecemos em quais casos é melhor usar um ou outro método.
Os processos de oxidação, combustão, halogenação, nitração, desidrogenação, decomposição são processos redox. Ao passar de uma classe de compostos orgânicos para outra e aumentar o grau de ramificação do esqueleto de carbono das moléculas de compostos dentro de uma classe separada, o grau de oxidação do átomo de carbono responsável pela capacidade redutora do composto muda. Substâncias orgânicas, cujas moléculas contêm átomos de carbono com valores máximos (- e +) de CO (-4, -3, +2, +3), entram em uma reação completa de queima de oxidação, mas são resistentes a leves e oxidantes de força média. Substâncias cujas moléculas contêm átomos de carbono no CO -1; 0; +1, são facilmente oxidados, suas habilidades de redução são próximas, então sua oxidação incompleta pode ser alcançada usando um dos agentes oxidantes conhecidos de força baixa e média. Essas substâncias podem apresentar natureza dual, atuando como agente oxidante, assim como é inerente às substâncias inorgânicas.
Alcanos

Alcenos
Os processos de oxidação dependem da estrutura do alceno e do meio reacional.

1. Quando os alcenos são oxidados com uma solução concentrada de permanganato de potássio KMnO4 em meio ácido (oxidação dura), as ligações σ e π se rompem com a formação de ácidos carboxílicos, cetonas e monóxido de carbono (IV). Esta reação é usada para determinar a posição da ligação dupla.

a) Se a dupla ligação estiver no final da molécula (por exemplo, no buteno-1), então um dos produtos da oxidação é o ácido fórmico, que é facilmente oxidado a dióxido de carbono e água:

b) Se na molécula de alceno o átomo de carbono na ligação dupla contém dois substituintes de carbono (por exemplo, na molécula de 2-metilbuteno-2), então durante sua oxidação é formada uma cetona, pois a transformação de tal átomo em um átomo do grupo carboxila é impossível sem quebrar a ligação C –C, relativamente estável nestas condições:

c) Se a molécula de alceno for simétrica e a ligação dupla estiver contida no meio da molécula, então apenas um ácido é formado durante a oxidação:

Uma característica da oxidação de alcenos, na qual os átomos de carbono na ligação dupla contêm dois radicais de carbono, é a formação de duas cetonas:


2. Em ambientes neutros ou levemente alcalinos, a oxidação é acompanhada pela formação de dióis (álcoois diídricos), e grupos hidroxila são ligados aos átomos de carbono entre os quais havia uma ligação dupla:

No decorrer desta reação, a cor violeta da solução aquosa de KMnO4 é descolorida. Portanto, é usado como uma reação qualitativa para alcenos (reação de Wagner).
3. A oxidação de alcenos na presença de sais de paládio (processo Wacker) leva à formação de aldeídos e cetonas:
2CH2=CH2 + O2 PdCl2/H2O → 2CH3-CO-H
Homólogos são oxidados no átomo de carbono menos hidrogenado: СH3-CH2-CH=CH2 + 1/2O2 PdCl2/H2O → CH3-CH2-CO-CH3 Alcinos
A oxidação do acetileno e seus homólogos ocorre dependendo do meio em que o processo ocorre.
a) Em meio ácido, o processo de oxidação é acompanhado pela formação de ácidos carboxílicos:

1 A reação é usada para determinar a estrutura de alcinos por produtos de oxidação:

2 Em meio neutro e levemente alcalino, a oxidação do acetileno é acompanhada pela formação dos oxalatos correspondentes (sais do ácido oxálico), e a oxidação dos homólogos é acompanhada pela quebra da ligação tripla e formação de sais de ácidos carboxílicos :

3 Para acetileno:
1) Em um ambiente ácido: H-C≡C-H KMnO4, H2SO4→ HOOC-COOH (ácido oxálico)
2) Em meio neutro ou alcalino: 3CH≡CH +8KMnO4 H2O→ 3KOOC-COOK oxalato de potássio +8MnO2↓+ 2KOH+ 2H2O
Arenos (benzeno e seus homólogos)
Ao oxidar arenos em meio ácido, deve-se esperar a formação de ácidos e, em meio alcalino, sais. Homólogos de benzeno com uma cadeia lateral (independentemente de seu comprimento) são oxidados por um forte agente oxidante a ácido benzóico no átomo de carbono α. Os homólogos do benzeno, quando aquecidos, são oxidados pelo permanganato de potássio em meio neutro para formar sais de potássio de ácidos aromáticos.
5C6H5–CH3 + 6KMnO4 + 9H2SO4 = 5C6H5COOH + 6MnSO4 + 3K2SO4 + 14H2O,
5C6H5–C2H5 + 12KMnO4 + 18H2SO4 = 5C6H5COOH + 5CO2 + 12MnSO4 + 6K2SO4 + 28H2O,
C6H5–CH3 + 2KMnO4 = C6H5COOK + 2MnO2 + KOH + H2O.
Enfatizamos que, se houver várias cadeias laterais em uma molécula de areno, em um meio ácido cada uma delas é oxidada em um átomo de carbono a para um grupo carboxila, resultando na formação de ácidos aromáticos polibásicos:

1) Em um ambiente ácido: С6H5-CH2-R KMnO4, H2SO4 → С6H5-COOH ácido benzóico + CO2
2) Em meio neutro ou alcalino: С6H5-CH2-R KMnO4, H2O/(OH) → С6H5-COOK + CO2
3) Oxidação de homólogos de benzeno com permanganato de potássio ou bicromato de potássio quando aquecido: С6H5-CH2-R KMnO4, H2SO4, t˚C → С6H5-COOHácido benzóico + R-COOH
4) Oxidação do cumeno com oxigênio na presença de um catalisador (método do cumeno para produção de fenol): C6H5CH(CH3)2 O2, H2SO4→ C6H5-OH fenol + CH3-CO-CH3 acetona
5C6H5CH(CH3)2 + 18KMnO4 + 27H2SO4 → 5C6H5COOH + 42H2O + 18MnSO4 + 10CO2 + K2SO4
Deve-se atentar para o fato de que durante a oxidação suave do estireno com o permanganato de potássio KMnO4 em meio neutro ou ligeiramente alcalino, a ligação π se rompe e o glicol (álcool diídrico) é formado. Como resultado da reação, a solução colorida de permanganato de potássio rapidamente se torna incolor e um precipitado marrom de óxido de manganês (IV) precipita. A oxidação com um agente oxidante forte - permanganato de potássio em meio ácido - leva à ruptura completa da dupla ligação e à formação de dióxido de carbono e ácido benzóico, enquanto a solução torna-se incolor.
C6H5−CH═CH2 + 2 KMnO4 + 3 H2SO4 → C6H5−COOH + CO2 + K2SO4 + 2 MnSO4 +4 H2O
Álcoois
Deve-se lembrar que:
1) álcoois primários são oxidados a aldeídos: 3CH3–CH2OH + K2Cr2O7 + 4H2SO4 = 3CH3–CHO + K2SO4 + Cr2(SO4)3 + 7H2O;
2) álcoois secundários são oxidados a cetonas:

3) para álcoois terciários, a reação de oxidação não é típica. Os álcoois terciários, em cujas moléculas não há átomo de hidrogênio no átomo de carbono contendo o grupo OH, não se oxidam em condições normais. Sob condições severas (sob a ação de agentes oxidantes fortes e em altas temperaturas), eles podem ser oxidados a uma mistura de ácidos carboxílicos de baixo peso molecular, ou seja, destruição do esqueleto de carbono. Quando o metanol é oxidado com uma solução acidificada de permanganato de potássio ou dicromato de potássio, CO2 é formado. Os álcoois primários durante a oxidação, dependendo das condições da reação, podem formar não apenas aldeídos, mas também ácidos. Por exemplo, a oxidação do etanol com dicromato de potássio no frio termina com a formação de ácido acético, e quando aquecido, acetaldeído:
3CH3–CH2OH + 2K2Cr2O7 + 8H2SO4 = 3CH3–COOH + 2K2SO4 + 2Cr2(SO4)3 + 11H2O,
3CH3–CH2OH + K2Cr2O7 + 4H2SO4
3CH3–CHO + K2SO4 + Cr2(SO4)3 + 7H2O
Lembre-se da influência do meio ambiente nos produtos das reações de oxidação do álcool, a saber: uma solução neutra quente de KMnO4 oxida o metanol a carbonato de potássio e outros álcoois a sais dos ácidos carboxílicos correspondentes:

Oxidação de glicóis
Os 1,2-glicóis são facilmente clivados sob condições suaves pela ação do ácido iódico. Dependendo da estrutura do glicol inicial, os produtos de oxidação podem ser aldeídos ou cetonas:
![]()
Se três ou mais grupos OH estiverem ligados a átomos de carbono adjacentes, então os átomos do meio ou do meio são convertidos em ácido fórmico quando oxidados com ácido clorídrico.
A oxidação de glicóis com permanganato de potássio em meio ácido ocorre de forma semelhante à clivagem oxidativa de alcenos e também leva à formação de ácidos ou cetonas, dependendo da estrutura do glicol inicial.
Aldeídos e cetonas
Os aldeídos são mais facilmente oxidados do que os álcoois nos ácidos carboxílicos correspondentes, não apenas sob a ação de agentes oxidantes fortes (oxigênio do ar, soluções acidificadas de KMnO4 e K2Cr2O7), mas também sob a ação de fracos (solução de amônia de óxido de prata ou cobre( II) hidróxido):
5CH3–CHO + 2KMnO4 + 3H2SO4 = 5CH3–COOH + 2MnSO4 + K2SO4 + 3H2O,
3CH3–CHO + K2Cr2O7 + 4H2SO4 = 3CH3–COOH + Cr2(SO4)3 + K2SO4 + 4H2O,
CH3–CHO + 2OH CH3–COONH4 + 2Ag + 3NH3 + H2O
Atenção especial!!! A oxidação do metanal com uma solução de amônia de óxido de prata leva à formação de carbonato de amônio em vez de ácido fórmico: HCHO + 4OH = (NH4)2CO3 + 4Ag + 6NH3 + 2H2O.
Como já mencionado, a oxidação da matéria orgânica é a introdução de oxigênio em sua composição e (ou) a eliminação de hidrogênio. A recuperação é o processo inverso (a introdução de hidrogênio e a eliminação de oxigênio). Dada a composição dos alcanos (СnH2n+2), podemos concluir que eles são incapazes de participar de reações de redução, mas podem participar de reações de oxidação.
Os alcanos são compostos com baixo grau de oxidação do carbono e, dependendo das condições da reação, podem ser oxidados para formar vários compostos.
Em temperaturas normais, os alcanos não reagem mesmo com agentes oxidantes fortes (H2Cr2O7, KMnO4, etc.). Quando introduzidos em uma chama aberta, os alcanos queimam. Ao mesmo tempo, em excesso de oxigênio, eles são completamente oxidados a CO2, onde o carbono tem o maior estado de oxidação de +4 e água. A combustão de hidrocarbonetos leva à quebra de todas as ligações C-C e C-H e é acompanhada pela liberação de uma grande quantidade de calor (reação exotérmica).
É geralmente aceito que o mecanismo de oxidação de alcanos inclui um processo de radical em cadeia, já que o oxigênio em si não é muito reativo, para abstrair um átomo de hidrogênio de um alcano é necessária uma partícula que iniciará a formação de um radical alquila que irá reage com o oxigênio, formando um radical peroxi. O radical peroxi pode então abstrair um átomo de hidrogênio de outra molécula de alcano para formar um hidroperóxido de alquila e um radical.

É possível oxidar alcanos com oxigênio atmosférico a 100-150 ° C na presença de um catalisador - acetato de manganês, essa reação é utilizada na indústria. A oxidação ocorre quando uma corrente de ar é soprada através da parafina fundida contendo um sal de manganês.
Porque como resultado da reação, forma-se uma mistura de ácidos, a seguir são separados da parafina não reagida por dissolução em álcali aquoso e, a seguir, neutralizados com ácido mineral.
Diretamente na indústria, este método é usado para obter ácido acético a partir de n-butano:
oxidação de alceno
As reações de oxidação de alcenos são divididas em dois grupos: 1) reações nas quais o esqueleto de carbono é preservado, 2) reações de destruição oxidativa do esqueleto de carbono da molécula ao longo da ligação dupla.
Reações de oxidação de alcenos com preservação do esqueleto de carbono
1. Epoxidação (reação de Prilezhaev)
Alcenos acíclicos e cíclicos, ao interagir com perácidos em meio apolar, formam epóxidos (oxiranos).
Além disso, os oxiranos podem ser obtidos pela oxidação de alcenos com hidroperóxidos na presença de catalisadores contendo molibdênio, tungstênio e vanádio:

O oxirano mais simples, o óxido de etileno, é produzido industrialmente pela oxidação do etileno com oxigênio na presença de prata ou óxido de prata como catalisador.

2. anti-hidroxilação (hidrólise de epóxidos)
A hidrólise ácida (ou alcalina) de epóxidos leva à abertura do ciclo do óxido com a formação de transdióis.

Na primeira etapa, ocorre a protonação do átomo de oxigênio do epóxido com a formação de um cátion oxônio cíclico, que se abre como resultado do ataque nucleofílico da molécula de água.
A abertura do anel epóxi catalisada por base também leva à formação de trans-glicóis.

3. sin-hidroxilação
Um dos métodos mais antigos para a oxidação de alcenos é a reação de Wagner (oxidação com permanganato de potássio). Inicialmente, durante a oxidação, forma-se um éster permanganato cíclico, que é hidrolisado a um diol vicinal:

Além da reação de Wagner, existe outro método para a sin-hidroxilação de alcenos sob a ação do óxido de ósmio (VIII), proposto por Krige. Sob a ação do tetróxido de ósmio sobre um alceno em éter ou dioxano, forma-se um precipitado preto do éster cíclico do ácido ósmico - osmato. No entanto, a adição de OsO4 à ligação múltipla é acentuadamente acelerada na piridina. O precipitado preto resultante de osmate é facilmente decomposto pela ação de uma solução aquosa de hidrossulfito de sódio:

O permanganato de potássio ou óxido de ósmio (VIII) oxida o alceno a cis-1,2-diol.
Clivagem oxidativa de alcenos
A clivagem oxidativa dos alcenos inclui reações de sua interação com o permanganato de potássio em ácido alcalino ou sulfúrico, bem como a oxidação com uma solução de trióxido de cromo em ácido acético ou dicromato de potássio e ácido sulfúrico. O resultado final de tais transformações é a divisão do esqueleto de carbono no local da dupla ligação e a formação de ácidos carboxílicos ou cetonas.
Alcenos monossubstituídos com uma ligação dupla terminal são clivados em um ácido carboxílico e dióxido de carbono:

Se ambos os átomos de carbono na ligação dupla contiverem apenas um grupo alquila, forma-se uma mistura de ácidos carboxílicos:

Mas se um alceno tetrasubstituído com uma ligação dupla é uma cetona:

A reação de ozonólise de alcenos adquiriu um significado preparativo muito maior. Por muitas décadas, essa reação serviu como o principal método para determinar a estrutura do alceno inicial. Esta reação é realizada passando uma corrente de uma solução de ozônio em oxigênio, uma solução de alceno em cloreto de metileno ou acetato de etila a -80 ... -100 ° C. O mecanismo desta reação foi estabelecido por Krige:


Ozonídeos são compostos instáveis que se decompõem com uma explosão. Existem duas formas de decomposição dos ozonídeos - oxidativa e redutora.
Durante a hidrólise, os ozonídeos são divididos em compostos carbonílicos e peróxido de hidrogênio. O peróxido de hidrogênio oxida aldeídos em ácidos carboxílicos - isso é decomposição oxidativa:

Muito mais importante é a divisão redutora dos ozonídeos. Os produtos da ozonólise são aldeídos ou cetonas, dependendo da estrutura do alceno inicial:
Além dos métodos acima, existe outro método proposto em 1955 por Lemieux:
No método Lemieux, não há procedimentos demorados para separar o dióxido de manganês, pois dióxido e manganato são novamente oxidados com periodato ao íon permanganato. Isso permite que apenas quantidades catalíticas de permanganato de potássio sejam usadas.
4.5. oxidação de alceno
É aconselhável dividir as reações de oxidação do alceno em dois grandes grupos: reações nas quais o esqueleto de carbono é preservado e reações de destruição oxidativa do esqueleto de carbono da molécula ao longo da dupla ligação. O primeiro grupo de reações inclui a epoxidação, bem como a hidroxilação, levando à formação de dióis vicinais (glicóis). No caso dos alcenos cíclicos, a hidroxilação forma transe- ou cis-dióis. Outro grupo inclui a ozonólise e reações de oxidação exaustiva de alcenos, levando à formação de vários tipos de compostos carbonílicos e ácidos carboxílicos.
4.5.a. Reações de oxidação de alcenos com preservação do esqueleto de carbono
1. Epoxidação (reação de N.A. Prilezhaev, 1909)
Alcenos acíclicos e cíclicos, ao interagir com perácidos (perácidos) RCOOOH em meio apolar e indiferente, formam epóxidos (oxiranos), portanto a reação em si é chamada de reação de epoxidação.
De acordo com a nomenclatura moderna IUPAC- um anel de três membros com um átomo de oxigênio é chamado oxirano.
A epoxidação de alcenos deve ser considerada como um processo síncrono e coordenado, que não envolve intermediários iônicos, como o cátion hidroxila OH+. Em outras palavras, a epoxidação de alcenos é um processo sin- adição de um átomo de oxigênio à ligação dupla com preservação completa da configuração dos substituintes na ligação dupla.
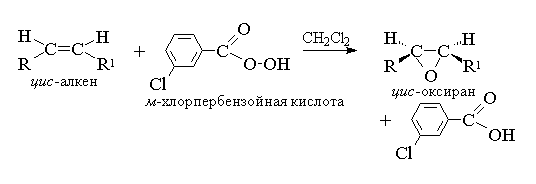

Para a epoxidação, foi proposto um mecanismo característico de processos concertados.

Como o ataque da ligação dupla pelo átomo de oxigênio do perácido é igualmente provável em ambos os lados do plano da ligação dupla, os oxiranos resultantes são ou meso-formas ou misturas de enantiômeros. Os seguintes perácidos são usados como agentes epoxidantes: perbenzóicos, m-clorperbenzóico, monoperftálico, peracético, trifluoroperacético e perfórmico. Os perácidos aromáticos são usados como reagentes individuais, enquanto os perácidos alifáticos - CH 3 CO 3 H, CF 3 CO 3 H e HCO 3 H não são isolados individualmente, mas são usados após sua formação na interação de peróxido de hidrogênio 30% ou 90% e o ácido carboxílico correspondente. Perbenzóico e m-ácido cloroperbenzóico é obtido pela oxidação de benzóico e m-ácido clorobenzóico com 70% de peróxido de hidrogênio em uma solução de ácido metanossulfônico ou de cloretos ácidos desses ácidos e peróxido de hidrogênio.


O ácido monoperftálico é obtido por um método semelhante a partir de anidrido ftálico e 30% de peróxido de hidrogênio.

Inicialmente, ácidos perbenzóicos ou monoperftálicos foram usados para obter oxiranos (epóxidos):

Atualmente, a epoxidação é mais usada m-ácido cloroperbenzóico. Ao contrário de outros perácidos, é estável durante o armazenamento por um longo período (até 1 ano) e é absolutamente seguro de manusear. Rendimentos de oxiranos obtidos por oxidação de alcenos acíclicos e cíclicos m-cloroperbenzóico em uma solução de cloreto de metileno, clorofórmio ou dioxano são geralmente bastante elevados.



Os perácidos são frequentemente gerados diretamente de uma mistura de reação de 90% de peróxido de hidrogênio e ácido carboxílico em cloreto de metileno.


Os alcenos com ligação dupla conjugada com um grupo carbonila ou outro substituinte aceitador são inativos e é melhor usar agentes oxidantes mais fortes para sua oxidação, como o ácido trifluoroperacético obtido do anidrido trifluoroacético e 90% de peróxido de hidrogênio em cloreto de metileno. O oxirano mais simples, o óxido de etileno, é produzido industrialmente pela oxidação do etileno com oxigênio na presença de prata como catalisador.

2. anti-Hidroxilação
O anel de três membros dos oxiranos é facilmente aberto sob a ação de uma grande variedade de reagentes nucleofílicos. Essas reações serão discutidas em detalhes na seção sobre éteres acíclicos e cíclicos. Aqui, apenas a hidrólise de oxiranos será considerada. A hidrólise de oxiranos é catalisada por ácidos e bases. Em ambos os casos, dióis vicinais, ou seja, glicóis, são formados. Durante a catálise ácida, na primeira etapa, ocorre a protonação do átomo de oxigênio do oxirano com a formação de um cátion oxônio cíclico, que se abre como resultado do ataque nucleofílico de uma molécula de água:

A etapa chave na abertura do anel, que determina a velocidade de todo o processo, é o ataque nucleofílico da água na forma protonada do oxirano. Do ponto de vista do mecanismo, esse processo é semelhante à abertura do íon bromônio durante o ataque nucleofílico do íon brometo ou outro agente nucleofílico. A partir dessas posições, o resultado estereoquímico deve ser a formação transe-glicóis na clivagem de epóxidos cíclicos. De fato, durante a hidrólise catalisada por ácido do óxido de ciclohexeno ou óxido de ciclopenteno, exclusivamente transe-1,2-dióis.

Assim, o processo de duas etapas de epoxidação do alceno seguido de hidrólise ácida do epóxido corresponde totalmente à reação anti-hidroxilação de alcenos.
Ambos os estágios anti A hidroxilação de alcenos pode ser combinada se o alceno for tratado com peróxido de hidrogênio aquoso a 30-70% em ácido fórmico ou trifluoroacético. Ambos os ácidos são fortes o suficiente para abrir o anel oxirano.

A abertura do anel oxirano, catalisada pela base, também leva à formação de transe-glicóis.

Portanto, o processo de dois estágios de epoxidação de alcenos seguido por hidrólise alcalina de epóxidos também é uma reação anti-hidroxilação de alcenos.
3. sin-Hidroxilação
Alguns sais e óxidos de metais de transição em estados de oxidação mais elevados são reagentes eficazes. sin-hidroxilação da ligação dupla de um alceno, quando ambos os grupos hidroxila estão ligados ao mesmo lado da ligação dupla. A oxidação de alcenos com permanganato de potássio é um dos métodos mais antigos sin-hidroxilação de dupla ligação - continua a ser amplamente utilizado, apesar de suas limitações inerentes. cis-1,2-ciclohexanodiol foi obtido pela primeira vez por V.V. Markovnikov em 1878 pela hidroxilação do ciclohexeno com uma solução aquosa de permanganato de potássio a 0 0 C.

Este método foi desenvolvido nas obras do cientista russo E.E. Wagner, portanto sin A hidroxilação de alcenos sob a ação de uma solução aquosa de permanganato de potássio é chamada de reação de Wagner. O permanganato de potássio é um forte agente oxidante que pode não apenas hidroxilar a ligação dupla, mas também clivar o diol vicinal resultante. Para evitar ao máximo a degradação dos glicóis, as condições de reação devem ser cuidadosamente controladas. Os rendimentos de glicol são geralmente baixos (30-60%). Os melhores resultados são obtidos pela hidroxilação de alcenos em um meio levemente alcalino (рН~8 9) a 0-5 0 С com uma solução aquosa diluída a 1% de KMnO 4 .


Inicialmente, quando os alcenos são oxidados com permanganato de potássio, forma-se um éster cíclico do ácido permangânico, que é imediatamente hidrolisado a um diol vicinal.

O éster cíclico do ácido permangânico não foi isolado como um intermediário, mas sua formação decorre de experimentos com permanganato de potássio 18 O marcado: ambos os átomos de oxigênio no glicol são marcados pela oxidação do alceno KMn 18 O 4 . Isso significa que ambos os átomos de oxigênio são transferidos do agente oxidante e não do solvente - água, o que está de acordo com o mecanismo proposto.
Outro método sin A -hidroxilação de alcenos sob a ação do óxido de ósmio (VIII) OsO 4 foi proposta por R. Krige em 1936. O tetróxido de ósmio é uma substância cristalina incolor, volátil, facilmente solúvel em éter, dioxano, piridina e outros solventes orgânicos. Quando o tetróxido de ósmio reage com alcenos em éter ou dioxano, forma-se um precipitado preto do éster cíclico do ácido ósmico - osmato, que pode ser facilmente isolado individualmente. A adição de OsO 4 à ligação dupla é acentuadamente acelerada em solução de piridina. A decomposição dos osmatos em glicóis vicinais é conseguida pela ação de uma solução aquosa de hidrossulfito de sódio ou sulfeto de hidrogênio.

Saídas do produto sin A -hidroxilação de alcenos neste método é muito maior do que quando se usa permanganato como agente oxidante. Uma vantagem importante do método de Krige é a ausência de produtos da clivagem oxidativa de alcenos, característica da oxidação do permanganato.


O tetróxido de ósmio é um reagente muito caro e difícil de obter, além de ser tóxico. Portanto, o óxido de ósmio (VIII) é usado na síntese de pequenas quantidades de substâncias de difícil acesso para obter o maior rendimento de diol. Para simplificar sin-hidroxilação de alcenos sob a ação de OsO 4 foi desenvolvida uma técnica que permite utilizar apenas quantidades catalíticas deste reagente. A hidroxilação de alcenos é realizada usando peróxido de hidrogênio na presença de OsO 4, por exemplo:

Para concluir esta seção, apresentamos as relações estereoquímicas entre o alceno cis- ou transe-configuração e configuração do diol vicinal resultante, que pode ser cis- ou transe-isômero, eritro- ou treo-forma, meso- ou D,L-forma dependendo dos substituintes no alceno:

Relações estereoquímicas semelhantes são observadas em outras reações sin- ou anti- adições de ligações múltiplas de hidrogênio, haletos de hidrogênio, água, halogênios, hidretos de boro e outros reagentes.