Sa mga reaksiyong redox, mga organikong sangkap mas madalas na nagpapakita sila ng mga katangian ng pagbabawas ng mga ahente, at ang kanilang mga sarili ay na-oxidized. Ang kadalian ng oksihenasyon ng mga organikong compound ay nakasalalay sa pagkakaroon ng mga electron kapag nakikipag-ugnayan sa ahente ng oxidizing. Ang lahat ng kilalang salik na nagdudulot ng pagtaas ng density ng elektron sa mga molekula ng mga organikong compound (halimbawa, mga positibong inductive at mesomeric na epekto) ay tataas ang kanilang kakayahang mag-oxidize at vice versa.
Ang pagkahilig ng mga organikong compound na mag-oxidize ay tumataas sa kanilang nucleophilicity, na tumutugma sa mga sumusunod na row:
Pagtaas ng nucleophilicity sa serye
Isaalang-alang natin mga reaksyon ng redox mga kinatawan ng pinakamahalagang klase organikong bagay na may ilang inorganikong oxidizing agent.
Oksihenasyon ng mga alkenes
Sa panahon ng banayad na oksihenasyon, ang mga alkenes ay na-convert sa glycols (dihydric alcohols). Ang mga nagpapababang atomo sa mga reaksyong ito ay mga atomo ng carbon na pinag-uugnay ng isang dobleng bono.
Ang reaksyon sa isang solusyon ng potassium permanganate ay nangyayari sa isang neutral o bahagyang alkalina na daluyan tulad ng sumusunod:
3C 2 H 4 + 2KMnO 4 + 4H 2 O → 3CH 2 OH–CH 2 OH + 2MnO 2 + 2KOH
Sa ilalim ng mas malubhang mga kondisyon, ang oksihenasyon ay humahantong sa pagkasira ng carbon chain sa double bond at pagbuo ng dalawang acids (sa isang malakas na alkaline na kapaligiran - dalawang salts) o isang acid at carbon dioxide (sa isang malakas na alkaline na kapaligiran - isang asin at isang carbonate):
1) 5CH 3 CH=CHCH 2 CH 3 + 8KMnO 4 + 12H 2 SO 4 → 5CH 3 COOH + 5C 2 H 5 COOH + 8MnSO 4 + 4K 2 SO 4 + 17H 2 O
2) 5CH 3 CH=CH 2 + 10KMnO 4 + 15H 2 SO 4 → 5CH 3 COOH + 5CO 2 + 10MnSO 4 + 5K 2 SO 4 + 20H 2 O
3) CH 3 CH=CHCH 2 CH 3 + 8KMnO 4 + 10KOH → CH 3 COOK + C 2 H 5 COOK + 6H 2 O + 8K 2 MnO 4
4) CH 3 CH=CH 2 + 10KMnO 4 + 13KOH → CH 3 COOK + K 2 CO 3 + 8H 2 O + 10K 2 MnO 4
Ang potassium dichromate sa isang sulfuric acid medium ay nag-oxidize ng mga alkenes na katulad ng mga reaksyon 1 at 2.
Sa panahon ng oksihenasyon ng mga alkenes, kung saan ang mga carbon atom sa double bond ay naglalaman ng dalawang carbon radical, dalawang ketone ang nabuo:


Alkyne oxidation
Ang mga alkynes ay nag-o-oxidize sa ilalim ng bahagyang mas malubhang mga kondisyon kaysa sa mga alkenes, kaya karaniwan silang nag-oxidize sa pamamagitan ng pagsira sa carbon chain sa triple bond. Tulad ng sa kaso ng mga alkenes, ang pagbabawas ng mga atom dito ay mga carbon atom na konektado sa pamamagitan ng maraming bono. Bilang resulta ng mga reaksyon, nabuo ang mga acid at carbon dioxide. Maaaring isagawa ang oksihenasyon gamit ang potassium permanganate o dichromate sa isang acidic na kapaligiran, halimbawa:
5CH 3 C≡CH + 8KMnO 4 + 12H 2 SO 4 → 5CH 3 COOH + 5CO 2 + 8MnSO 4 + 4K 2 SO 4 + 12H 2 O

Ang acetylene ay maaaring ma-oxidize ng potassium permanganate sa isang neutral na kapaligiran sa potassium oxalate:
3CH≡CH +8KMnO 4 → 3KOOC –COOK +8MnO 2 +2KOH +2H 2 O
Sa isang acidic na kapaligiran, ang oksihenasyon ay nagpapatuloy sa oxalic acid o carbon dioxide:
5CH≡CH +8KMnO 4 +12H 2 SO 4 → 5HOOC –COOH +8MnSO 4 +4K 2 SO 4 +12H 2 O
CH≡CH + 2KMnO 4 +3H 2 SO 4 → 2CO 2 + 2MnSO 4 + 4H 2 O + K 2 SO 4
Oxidation ng benzene homologues
Ang Benzene ay hindi nag-oxidize kahit na sa ilalim ng medyo malupit na mga kondisyon. Ang mga Benzene homologue ay maaaring ma-oxidized sa isang solusyon ng potassium permanganate sa isang neutral na kapaligiran sa potassium benzoate:
C 6 H 5 CH 3 + 2KMnO 4 → C 6 H 5 COOK + 2MnO 2 + KOH + H 2 O
C 6 H 5 CH 2 CH 3 + 4KMnO 4 → C 6 H 5 COOK + K 2 CO 3 + 2H 2 O + 4MnO 2 + KOH
Ang oksihenasyon ng mga benzene homologue na may potassium dichromate o permanganate sa isang acidic na kapaligiran ay humahantong sa pagbuo ng benzoic acid.
5C 6 H 5 CH 3 +6KMnO 4 +9 H 2 SO 4 → 5C 6 H 5 COOH+6MnSO 4 +3K 2 SO 4 + 14H 2 O
5C 6 H 5 –C 2 H 5 + 12KMnO 4 + 18H 2 SO 4 → 5C 6 H 5 COOH + 5CO 2 + 12MnSO 4 + 6K 2 SO 4 + 28H 2 O



Oksihenasyon ng mga alkohol
Ang direktang produkto ng oksihenasyon ng mga pangunahing alkohol ay aldehydes, at ang mga produkto ng oksihenasyon ng pangalawang alkohol ay mga ketone.
Ang mga aldehyde na nabuo sa panahon ng oksihenasyon ng mga alkohol ay madaling na-oxidized sa mga acid, samakatuwid ang mga aldehydes mula sa mga pangunahing alkohol ay nakuha sa pamamagitan ng oksihenasyon na may potassium dichromate sa isang acidic na daluyan sa kumukulong punto ng aldehyde. Kapag ang aldehydes ay sumingaw, wala silang oras upang mag-oxidize.
3C 2 H 5 OH + K 2 Cr 2 O 7 + 4H 2 SO 4 → 3CH 3 CHO + K 2 SO 4 + Cr 2 (SO 4) 3 + 7H 2 O
Sa labis na ahente ng oxidizing (KMnO 4, K 2 Cr 2 O 7) sa anumang kapaligiran, ang mga pangunahing alkohol ay na-oxidized sa mga carboxylic acid o sa kanilang mga asin, at ang mga pangalawang alkohol ay na-oxidized sa mga ketone.
5C 2 H 5 OH + 4KMnO 4 + 6H 2 SO 4 → 5CH 3 COOH + 4MnSO 4 + 2K 2 SO 4 + 11H 2 O
3CH 3 –CH 2 OH + 2K 2 Cr 2 O 7 + 8H 2 SO 4 → 3CH 3 –COOH + 2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O
Ang mga tertiary alcohol ay hindi nag-oxidize sa ilalim ng mga kundisyong ito, ngunit ang methyl alcohol ay na-oxidize sa carbon dioxide.

Ang dihydric alcohol, ethylene glycol HOCH 2 –CH 2 OH, kapag pinainit sa isang acidic na kapaligiran na may solusyon ng KMnO 4 o K 2 Cr 2 O 7, ay madaling na-oxidized sa oxalic acid, at sa isang neutral na kapaligiran sa potassium oxalate.
5CH 2 (OH) – CH 2 (OH) + 8КMnO 4 +12H 2 SO 4 → 5HOOC –COOH +8MnSO 4 +4К 2 SO 4 +22Н 2 О
3CH 2 (OH) – CH 2 (OH) + 8KMnO 4 → 3KOOC –LUTO +8MnO 2 +2KOH +8H 2 O
Oxidation ng aldehydes at ketones
Ang mga aldehydes ay medyo malakas na mga ahente ng pagbabawas, at samakatuwid ay madaling na-oxidize ng iba't ibang mga ahente ng oxidizing, halimbawa: KMnO 4, K 2 Cr 2 O 7, OH, Cu(OH) 2. Ang lahat ng mga reaksyon ay nangyayari kapag pinainit:
3CH 3 CHO + 2KMnO 4 → CH 3 COOH + 2CH 3 COOK + 2MnO 2 + H 2 O
3CH 3 CHO + K 2 Cr 2 O 7 + 4H 2 SO 4 → 3CH 3 COOH + Cr 2 (SO 4) 3 + 7H 2 O
CH 3 CHO + 2KMnO 4 + 3KOH → CH 3 COOK + 2K 2 MnO 4 + 2H 2 O
5CH 3 CHO + 2KMnO 4 + 3H 2 SO 4 → 5CH 3 COOH + 2MnSO 4 + K 2 SO 4 + 3H 2 O
CH 3 CHO + Br 2 + 3NaOH → CH 3 COONa + 2NaBr + 2H 2 O

"salamin na pilak" ang reaksyon
Sa isang ammonia solution ng silver oxide, ang mga aldehydes ay na-oxidized sa mga carboxylic acid, na sa isang ammonia solution ay nagbibigay ng mga ammonium salts (ang "silver mirror" na reaksyon):
CH 3 CH=O + 2OH → CH 3 COONH 4 + 2Ag + H 2 O + 3NH 3
CH 3 –CH=O + 2Cu(OH) 2 → CH 3 COOH + Cu 2 O + 2H 2 O
Ang formic aldehyde (formaldehyde) ay karaniwang na-oxidized sa carbon dioxide:
5HCOH + 4KMnO4 (kubo) + 6H 2 SO 4 → 4MnSO 4 + 2K 2 SO 4 + 5CO 2 + 11H 2 O
3CH 2 O + 2K 2 Cr 2 O 7 + 8H 2 SO 4 → 3CO 2 +2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O
HCHO + 4OH → (NH 4) 2 CO 3 + 4Ag↓ + 2H 2 O + 6NH 3
HCOH + 4Cu(OH) 2 → CO 2 + 2Cu 2 O↓+ 5H 2 O
Ang mga ketone ay na-oxidize sa ilalim ng malupit na mga kondisyon sa pamamagitan ng malakas na mga ahente ng oxidizing na may pagkaputol ng mga C-C bond at nagbibigay ng mga pinaghalong acid:

Mga carboxylic acid. Kabilang sa mga acid, ang formic at oxalic acid ay may malakas na pagbabawas ng mga katangian, na nag-oxidize sa carbon dioxide.
HCOOH + HgCl 2 =CO 2 + Hg + 2HCl
HCOOH+ Cl 2 = CO 2 +2HCl
HOOC-COOH+ Cl 2 =2CO 2 +2HCl
Formic acid, bilang karagdagan sa mga acidic na katangian, ay nagpapakita rin ng ilang mga katangian ng aldehydes, sa partikular, pagbabawas ng mga katangian. Kasabay nito, ito ay na-oxidized sa carbon dioxide. Halimbawa:
2KMnO4 + 5HCOOH + 3H2SO4 → K2SO4 + 2MnSO4 + 5CO2 + 8H2O
Kapag pinainit ng malakas na dewatering agent (H2SO4 (conc.) o P4O10) ito ay nabubulok:
HCOOH →(t)CO + H2O
Catalytic oxidation ng alkanes:

Catalytic oxidation ng alkenes:

Oksihenasyon ng mga phenol:


Ang mga alkynes na may non-terminal triple bond ay nagsisilbing potensyal na mapagkukunan para sa synthesis ng 1,2-diketones sa ilalim ng pagkilos ng isang angkop na ahente ng oxidizing. Gayunpaman, ang isang unibersal na reagent ay hindi pa natagpuan na nagiging sanhi ng oksihenasyon ng isang triple carbon-carbon bond sa isang 1,2-dicarbonyl group. Ang RuO 4, ruthenium (VIII) oxide, na iminungkahi para sa layuning ito ay masyadong mahal at kadalasang nagiging sanhi ng karagdagang pagkasira ng oxidative ng 1,2-diketone sa mga carboxylic acid. Kapag ang disubstituted acetylenes ay nakikipag-ugnayan sa mga malakas na ahente ng oxidizing tulad ng potassium permanganate, tanging sa isang ganap na neutral na kapaligiran sa pH 7-8 sa 0 °C maaaring ihinto ang oksihenasyon sa yugto ng pagbuo ng α-diketone. Halimbawa, ang stearolic acid sa pH 7.5 ay na-oxidize sa α-diketone. Sa karamihan ng mga kaso, ang oksihenasyon ay sinamahan ng cleavage ng triple bond upang bumuo ng mga carboxylic acid:

Ang ani ng mga produkto mula sa oxidative na pagkasira ng mga alkynes ay maliit, at ang reaksyong ito ay hindi gumaganap ng isang makabuluhang papel sa organic synthesis. Ito ay ginagamit lamang upang patunayan ang istraktura ng natural na nagaganap na acetylene acid na matatagpuan sa mga dahon ng mga tropikal na halaman sa Central America. Sa panahon ng pagkasira ng oxidative nito, dalawang acid ang nahiwalay - lauric at adipic. Nangangahulugan ito na ang parent acid ay 6-octadecynic acid na may normal na carbon skeleton ng labimpitong carbon atoms:
Ang mas mahalaga ay ang oxidative coupling ng alkynes-1, na na-catalyze ng mga tansong asin (reaksyon ng Glaser-Eglinton). Noong 1870, natuklasan ni Glaser na ang isang suspensyon ng tanso (I) acetylide sa alkohol ay na-oxidize ng atmospheric oxygen upang bumuo ng 1,3-diynes:

Para sa oksihenasyon ng copper(I) acetylenides, ang potassium hexacyanoferrate(III) K3 sa DME o DMF ay mas epektibo bilang isang oxidizing agent. Noong 1959, iminungkahi ni Eglinton ang isang mas maginhawang pagbabago ng oxidative condensation ng alkynes. Ang alkyne ay na-oxidized na may tanso (II) acetate sa isang pyridine solution sa 60-70 °C. Ang Eglinton modification ay napatunayang lubhang kapaki-pakinabang para sa synthesis ng macrocyclic polyynes mula sa ,-diynes. Bilang isang paglalarawan, ipinakita namin ang synthesis ng dalawang cyclopolyin sa panahon ng oxidative condensation ng hexadiine-1,5 (F. Sondheimer, 1960):

Ang isa sa mga polyine ay isang produkto ng cyclotrimerization, ang isa ay isang produkto ng cyclotetramerization ng orihinal na hesadiin-1,5. Ang trimer ay nagsisilbing panimulang reagent para sa synthesis ng aromatic -annulene (para sa karagdagang impormasyon sa annulenes, tingnan ang Kabanata 12). Katulad nito, sa ilalim ng parehong mga kondisyon ng nonadiine-1,8, ang dimer nito ay nakuha - 1,3,10,12-cyclooctadecatetraene, kasama ang isang trimer, tetramer at pentamer:

Upang makakuha ng hindi simetriko diynes, ang condensation ng haloacetylenes na may alkyne-1 (terminal alkyne) sa pagkakaroon ng tanso (I) na mga asin at isang pangunahing amine ay ginagamit (kumbinasyon ayon kay Kadio-Khodkevich, 1957):

Ang panimulang bromoalkynes ay nakuha sa pamamagitan ng pagkilos ng sodium hypobromite sa alkynes-1 o mula sa lithium at bromine acetylenides:
Ang organocopper derivative ng terminal alkyne ay direktang nabuo sa reaction mixture ng Cu 2 Cl 2 at alkyne-1.
6.3.4. Mga reaksyon ng electrophilic na karagdagan sa isang triple bond
Ang mga electrophilic na reaksyon ng karagdagan sa isang triple bond ay kabilang sa mga pinakakaraniwang at mahalagang reaksyon ng alkynes. Sa kaibahan sa electrophilic na karagdagan sa mga alkenes, ang sintetikong aplikasyon ng malaking pangkat ng mga reaksyon na ito ay nauuna sa pagbuo ng mga teoretikal na ideya tungkol sa mekanismo nito. Gayunpaman, sa nakalipas na dalawampung taon ang sitwasyon ay nagbago nang malaki at sa kasalukuyan ito ay isa sa mabilis na umuunlad na mga lugar ng pisikal na organikong kimika. Ang HOMO ng isang alkyne ay matatagpuan na mas mababa kaysa sa HOMO ng isang alkene (Kabanata 2), at ang pangyayaring ito ay tumutukoy sa karamihan ng mga kaso ng isang mas mababang rate ng pagdaragdag ng isang electrophilic agent sa isang alkyne kumpara sa isang alkene. Ang isa pang kadahilanan na tumutukoy sa pagkakaiba sa reaktibiti ng mga alkynes at alkenes sa mga reaksyon ng pagdaragdag ng electrophilic ay ang relatibong katatagan ng mga intermediate na nagreresulta mula sa pagdaragdag ng isang electrophilic species sa triple at double bond. Kapag ang isang electrophilic H + o E + species ay nakakabit sa isang double bond, isang cyclic o open carbocation ay nabuo (Kabanata 5). Ang pagdaragdag ng H+ o E+ sa isang triple bond ay nagreresulta sa pagbuo ng isang bukas o cyclic vinyl cation. Sa isang linear na bukas na vinyl cation, ang gitnang carbon atom ay matatagpuan sa sp-hybrid state, habang bakante r-orbital ay orthogonal sa -bond. Since sp-ang hybrid carbon atom ng vinyl cation ay may mas mataas na electronegativity kumpara sa sp 2-hybrid atom ng alkyl cation, ang vinyl cation ay dapat na hindi gaanong matatag kumpara sa alkyl cation:

Ang data mula sa quantum mechanical calculations, pati na rin ang thermodynamic data para sa gas phase na nakuha gamit ang high-pressure mass spectrometry at cyclotron resonance spectroscopy, ay ganap na sumasang-ayon sa mga pagsasaalang-alang na ito. Sa mesa Ang 6.3 ay nagpapakita ng thermodynamic data para sa pagbuo ng isang bilang ng mga carbocation at hydrocarbon na nauugnay sa gas phase sa 25 °C.
|
Carbocation |
Δ N f ˚ kcal/mol |
|
| |
|
| |
|
|
Mula sa datos na ipinakita sa talahanayan. 6.3, ito ay sumusunod na ang vinyl cation ay 47 kcal/mol na hindi gaanong matatag kaysa sa ethyl cation na naglalaman ng parehong bilang ng mga atomo. Ang parehong konklusyon ay maaaring makuha mula sa enthalpy ng ionization sa gas phase CH 3 CH 2 Cl at CH 2 =CHCl:
Madaling makita na ang kumbinasyon ng parehong mga kadahilanan - ang mas mataas na enerhiya ng vinyl cation at ang mababang HOMO ng alkyne - ay kumakatawan sa isang mas mababang reaktibiti ng mga alkynes kumpara sa mga alkenes sa mga reaksyon ng pagdaragdag ng electrophilic. Sa mesa Ang 6.4 ay naglalaman ng comparative data sa pagdaragdag ng mga halogens, sulfene at selenyl chlorides, trifluoroacetic acid at tubig sa iba't ibang alkenes at alkynes na hindi naglalaman ng anumang nag-activate o nagde-deactivate na functional group.
Talahanayan 6.4
Mga paghahambing na katangian ng alkynes at alkenes
sa mga reaksyon ng pagdaragdag ng electrophilic
|
Mga substrate |
K alkene/K alkyne |
|
|
Bromination sa acetic acid |
CH 2 CH 2 /HCCH C 4 H 9 CH = CH 2 / C 4 H 9 CCH C 6 H 5 CH=CH 2 /C 6 H 5 CCH | |
|
Chlorination sa acetic acid |
C 6 H 5 CH=CH 2 /C 6 H 5 CCH C 4 H 9 CH = CH 2 / C 6 H 5 CCH С 2 Н 5 С=СНС 2 Н 5 /С 2 Н 5 ССС 2 Н 5 | |
|
Pagdaragdag ng 4-chlorophenylsulfene chloride n-ClС 6 H 4 SeCl |
CH 2 =CH 2 /HCCH C 4 H 9 CH = CH 2 / C 4 H 9 CCH C 6 H 5 CH=CH 2 /C 6 H 5 CCH | |
|
Pagdaragdag ng phenylselenium chloride C 6 H 5 SeCl |
CH 2 =CH 2 /HCCH C 4 H 9 CH = CH 2 / C 4 H 9 CCH C 6 H 5 CH=CH 2 /C 6 H 5 CCH | |
|
Pagdaragdag ng trifluoroacetic acid |
C 4 H 9 CH = CH 2 / C 4 H 9 CCH C 6 H 5 CH=CH 2 /C 6 H 5 CCH C 2 H 5 CH = CH 2 / C 2 H 5 CCH | |
|
Acid-catalyzed hydration |
C 4 H 9 CH = CH 2 / C 4 H 9 CCH С 2 Н 5 СН=СНС 2 Н 5 /С 2 Н 5 ССС 2 Н 5 C 6 H 5 CH=CH 2 /C 6 H 5 CCH |
Mula sa mga datos na ito ay sumusunod na ang pagdaragdag lamang ng mga acidic na ahente at tubig sa triple at dobleng mga bono ay nangyayari sa magkatulad na mga rate. Ang pagdaragdag ng mga halogens, sulfene chlorides at ilang iba pang reagents sa alkenes ay nangyayari nang 10 2 - 10 5 beses na mas mabilis kaysa sa mga alkynes. Nangangahulugan ito na ang mga hydrocarbon na naglalaman ng di-conjugated na triple at double bond ay piling nagdaragdag ng mga reagents na ito sa double bond, halimbawa:

Ang data sa comparative hydration ng alkynes at alkenes ay dapat tratuhin nang may pag-iingat dahil ang hydration ng alkynes ay nangangailangan ng mercury(II) ion catalysis, na hindi epektibo para sa pagdaragdag ng tubig sa double bond. Samakatuwid, ang data sa hydration ng triple at double bond, mahigpit na nagsasalita, ay hindi maihahambing.
Ang pagdaragdag ng mga halogens, hydrogen halides, sulfene chlorides at iba pang mga electrophilic agent ay maaaring isagawa sa mga hakbang, na madaling ilarawan gamit ang mga sumusunod na halimbawa:



Ang pagkahilig ng mga organikong compound na mag-oxidize ay nauugnay sa pagkakaroon ng maraming mga bono, functional group, at hydrogen atoms sa carbon atom na naglalaman ng functional group. Ang sunud-sunod na oksihenasyon ng mga organikong sangkap ay maaaring kinakatawan bilang ang sumusunod na kadena ng mga pagbabagong-anyo:
Saturated hydrocarbon → Unsaturated hydrocarbon → Alcohol → Aldehyde (ketone) → Carboxylic acid → CO2 + H2O
Ang genetic na relasyon sa pagitan ng mga klase ng mga organikong compound ay kinakatawan dito bilang isang serye ng mga redox na reaksyon na nagsisiguro ng paglipat mula sa isang klase ng mga organikong compound patungo sa isa pa. Nakumpleto ito ng mga produkto ng kumpletong oksihenasyon (pagkasunog) ng anumang kinatawan ng mga klase ng mga organikong compound. Ang pagdepende sa kakayahan ng redox ng isang organikong sangkap sa istraktura nito: Ang tumaas na tendensya ng mga organikong compound na mag-oxidize ay dahil sa pagkakaroon ng mga sangkap sa molekula: maraming mga bono (ito ang dahilan kung bakit ang mga alkenes, alkynes, alkadienes ay napakadaling na-oxidize); ilang mga functional na grupo na madaling ma-oxidize (–-SH, –OH (phenolic at alcoholic), – NH2; activated alkyl group na matatagpuan katabi ng maraming bond.
Halimbawa, ang propene ay maaaring ma-oxidize sa unsaturated aldehyde acrolein na may atmospheric oxygen sa pagkakaroon ng singaw ng tubig sa bismuth-molybdenum catalysts.
H2C═CH−CH3 → H2C═CH−COH
At din ang oksihenasyon ng toluene sa benzoic acid na may potassium permanganate sa isang acidic na kapaligiran. 5C6H5CH3 +6KMnO4 + 9H2SO4 → 5C6H5COOH + 3K2SO4 + 6MnSO4 +14H2O
ang pagkakaroon ng hydrogen atoms sa isang carbon atom na naglalaman ng functional group. Ang isang halimbawa ay ang reaktibiti sa mga reaksyon ng oksihenasyon ng pangunahin, pangalawa at tertiary na alkohol sa pamamagitan ng reaktibidad ng oksihenasyon.
Sa kabila ng katotohanan na sa panahon ng anumang reaksyon ng redox ay nangyayari ang parehong oksihenasyon at pagbawas, ang mga reaksyon ay inuri depende sa kung ano ang direktang nangyayari sa organic compound (kung ito ay na-oxidized, pinag-uusapan natin ang proseso ng oksihenasyon, kung ito ay nabawasan, pinag-uusapan natin ang proseso ng pagbabawas. ).
Kaya, sa reaksyon ng ethylene na may potassium permanganate, ang ethylene ay ma-oxidized, at ang potassium permanganate ay mababawasan. Ang reaksyon ay tinatawag na ethylene oxidation.
Kapag pinag-aaralan ang mga comparative na katangian ng inorganic at organic compounds, naging pamilyar kami sa paggamit ng oxidation state (s.o.) (sa organic chemistry, pangunahin ang carbon) at mga pamamaraan para sa pagtukoy nito:
1) pagkalkula ng average na s.o. carbon sa isang molekula ng organikong bagay: -8/3 +1 C3 H8 Ang pamamaraang ito ay makatwiran kung sa panahon ng reaksyon sa organikong bagay lahat ng mga bono ng kemikal ay nawasak (pagkasunog, kumpletong pagkabulok).
2) kahulugan ng s.o. bawat carbon atom:
Sa kasong ito, ang estado ng oksihenasyon ng anumang carbon atom sa isang organikong tambalan ay katumbas ng algebraic na kabuuan ng mga bilang ng lahat ng mga bono na may mga atomo ng mas maraming electronegative na elemento, na isinasaalang-alang sa "+" sign sa carbon atom, at ang bilang ng mga bono na may mga atomo ng hydrogen (o isa pang mas electropositive na elemento), na isinasaalang-alang kasama ang sign na "-" sa carbon atom. Sa kasong ito, ang mga bono sa mga kalapit na carbon atom ay hindi isinasaalang-alang. Bilang isang simpleng halimbawa, alamin natin ang estado ng oksihenasyon ng carbon sa isang molekula ng methanol. Ang isang carbon atom ay konektado sa tatlong hydrogen atoms (ang mga bond na ito ay binibilang na may “–” sign), at isang bond ay konektado sa isang oxygen atom (ito ay binibilang na may “+” sign). Nakukuha namin ang: -3 + 1 = -2 Kaya, ang estado ng oksihenasyon ng carbon sa methanol ay -2. Ang kinakalkula na antas ng oksihenasyon ng carbon, kahit na isang kondisyon na halaga, ay nagpapahiwatig ng likas na katangian ng pagbabago sa density ng elektron sa molekula, at ang pagbabago nito bilang resulta ng reaksyon ay nagpapahiwatig ng proseso ng redox na nagaganap. Linawin natin kung anong mga kaso ang mas mahusay na gumamit ng isang paraan o iba pa.
Ang mga proseso ng oxidation, combustion, halogenation, nitration, dehydrogenation, at decomposition ay inuri bilang redox process. Kapag lumipat mula sa isang klase ng mga organikong compound patungo sa isa pa at pinapataas ang antas ng pagsasanga ng carbon skeleton ng mga molekula ng mga compound sa loob ng isang hiwalay na klase, ang antas ng oksihenasyon ng carbon atom na responsable para sa pagbabawas ng kakayahan ng compound ay nagbabago. Ang mga organikong sangkap, ang mga molekula na naglalaman ng mga carbon atom na may pinakamataas na (- at +) CO na mga halaga (-4, -3, +2, +3), ay pumapasok sa isang kumpletong reaksyon ng oksihenasyon-pagkasunog, ngunit lumalaban sa banayad at medium-strength oxidizing agent . Mga sangkap na ang mga molekula ay naglalaman ng mga carbon atom sa CO -1; 0; +1, madaling mag-oxidize, ang kanilang mga kakayahan sa pagbabawas ay malapit na, kaya ang kanilang hindi kumpletong oksihenasyon ay maaaring makamit gamit ang isa sa mga kilalang oxidizing agent na mababa at katamtamang lakas. Ang mga sangkap na ito ay maaaring magpakita ng dalawahang katangian, na kumikilos bilang isang ahente ng oxidizing, tulad ng likas sa mga di-organikong sangkap.
Alkanes

Alkenes
Ang mga proseso ng oksihenasyon ay nakasalalay sa istruktura ng alkene at sa kapaligiran ng reaksyon.

1. Kapag ang mga alkenes ay na-oxidized na may concentrated na solusyon ng potassium permanganate KMnO4 sa isang acidic na kapaligiran (hard oxidation), ang σ- at π-bond ay nasisira upang bumuo ng mga carboxylic acid, ketone at carbon monoxide (IV). Ang reaksyong ito ay ginagamit upang matukoy ang posisyon ng double bond.

a) Kung ang dobleng bono ay nasa dulo ng molekula (halimbawa, sa butene-1), kung gayon ang isa sa mga produkto ng oksihenasyon ay formic acid, na madaling na-oxidize sa carbon dioxide at tubig:

b) Kung sa isang alkene molecule ang carbon atom sa double bond ay naglalaman ng dalawang carbon substituents (halimbawa, sa 2-methylbutene-2 molecule), pagkatapos ay sa panahon ng oksihenasyon nito ay nabuo ang isang ketone, dahil ang pagbabago ng naturang atom sa isang carboxyl group atom ay imposible nang hindi nasisira ang C-C-bond, medyo matatag sa ilalim ng mga kondisyong ito:

c) Kung ang molekula ng alkene ay simetriko at ang dobleng bono ay nakapaloob sa gitna ng molekula, kung gayon isang acid lamang ang nabuo sa panahon ng oksihenasyon:

Ang isang tampok ng oksihenasyon ng mga alkenes, kung saan ang mga carbon atom sa double bond ay naglalaman ng dalawang carbon radical, ay ang pagbuo ng dalawang ketones:


2. Sa neutral o bahagyang alkaline na media, ang oksihenasyon ay sinamahan ng pagbuo ng mga diol (dihydric alcohols), at ang mga hydroxyl group ay idinagdag sa mga carbon atom na iyon kung saan nagkaroon ng double bond:

Sa panahon ng reaksyong ito, ang kulay violet ng may tubig na solusyon ng KMnO4 ay nagiging kupas. Samakatuwid, ito ay ginagamit bilang isang qualitative na reaksyon para sa mga alkenes (Wagner reaction).
3. Ang oksihenasyon ng mga alkenes sa pagkakaroon ng mga palladium salts (proseso ng Wacker) ay humahantong sa pagbuo ng mga aldehydes at ketones:
2CH2=CH2 + O2 PdCl2/H2O → 2 CH3-CO-H
Ang mga homolog ay na-oxidized sa hindi gaanong hydrogenated na carbon atom: CH3-CH2-CH=CH2 + 1/2O2 PdCl2/H2O → CH3- CH2-CO-CH3 Alkynes
Ang oksihenasyon ng acetylene at ang mga homologue nito ay nangyayari depende sa kapaligiran kung saan nagaganap ang proseso.
a) Sa isang acidic na kapaligiran, ang proseso ng oksihenasyon ay sinamahan ng pagbuo ng mga carboxylic acid:

1 Ang reaksyon ay ginagamit upang matukoy ang istraktura ng mga alkynes batay sa kanilang mga produkto ng oksihenasyon:

2 Sa neutral at bahagyang alkaline na kapaligiran, ang oksihenasyon ng acetylene ay sinamahan ng pagbuo ng kaukulang mga oxalate (oxalic acid salts), at ang oksihenasyon ng mga homologue ay sinamahan ng pagkalagot ng triple bond at pagbuo ng mga carboxylic acid salts:

3 Para sa acetylene:
1) Sa isang acidic na kapaligiran: H-C≡C-H KMnO4, H2SO4→ HOOC-COOH (oxalic acid)
2) Sa isang neutral o alkaline na kapaligiran: 3CH≡CH +8KMnO4 H2O→ 3KOOC-COOK potassium oxalate +8MnO2↓+ 2KOH+ 2H2O
Arenas (benzene at mga homologue nito)
Kapag ang mga arene ay na-oxidized sa isang acidic na kapaligiran, dapat asahan ng isa ang pagbuo ng mga acid, at sa isang alkaline na kapaligiran - mga asing-gamot. Ang mga Benzene homologue na may isang side chain (anuman ang haba nito) ay na-oxidize ng isang malakas na oxidizing agent sa benzoic acid sa α-carbon atom. Ang mga Benzene homologue, kapag pinainit, ay na-oxidized ng potassium permanganate sa isang neutral na kapaligiran upang bumuo ng mga potassium salt ng mga aromatic acid.
5C6H5–CH3 + 6KMnO4 + 9H2SO4 = 5C6H5COOH + 6MnSO4 + 3K2SO4 + 14H2O,
5C6H5–C2H5 + 12KMnO4 + 18H2SO4 = 5C6H5COOH + 5CO2 + 12MnSO4 + 6K2SO4 + 28H2O,
C6H5–CH3 + 2KMnO4 = C6H5COOK + 2MnO2 + KOH + H2O.
Binibigyang-diin namin na kung mayroong maraming mga side chain sa isang molekula ng arene, kung gayon sa isang acidic na kapaligiran, ang bawat isa sa kanila ay na-oxidized sa a-carbon atom sa isang carboxyl group, na nagreresulta sa pagbuo ng polybasic aromatic acid:

1) Sa isang acidic na kapaligiran: C6H5-CH2-R KMnO4, H2SO4→ C6H5-COOH benzoic acid + CO2
2) Sa isang neutral o alkaline na kapaligiran: C6H5-CH2-R KMnO4, H2O/(OH)→ C6H5-COOK + CO2
3) Oxidation ng benzene homologues na may potassium permanganate o potassium dichromate kapag pinainit: C6H5-CH2-R KMnO4, H2SO4, t˚C→ C6H5-COOHbenzoic acid + R-COOH
4) Oxidation ng cumene na may oxygen sa pagkakaroon ng isang katalista (paraan ng cumene para sa paggawa ng phenol): C6H5CH(CH3)2 O2, H2SO4→ C6H5-OH phenol + CH3-CO-CH3 acetone
5C6H5CH(CH3)2 + 18KMnO4 + 27H2SO4 → 5C6H5COOH + 42H2O + 18MnSO4 + 10CO2 + K2SO4
Dapat pansinin na sa panahon ng banayad na oksihenasyon ng styrene na may potassium permanganate KMnO4 sa isang neutral o bahagyang alkaline na daluyan, ang π bond ay nasira at ang glycol (dihydric alcohol) ay nabuo. Bilang resulta ng reaksyon, ang may kulay na solusyon ng potassium permanganate ay mabilis na nagiging kupas at isang kayumangging namuo ng manganese (IV) oxide ay namuo. Ang oksihenasyon na may malakas na ahente ng oxidizing - potassium permanganate sa isang acidic na kapaligiran - ay humahantong sa kumpletong pagkalagot ng double bond at pagbuo ng carbon dioxide at benzoic acid, at ang solusyon ay nagiging kupas.
C6H5−CH═CH2 + 2 KMnO4 + 3 H2SO4 → C6H5−COOH + CO2 + K2SO4 + 2 MnSO4 +4 H2O
Mga alak
Dapat tandaan na:
1) ang mga pangunahing alkohol ay na-oxidized sa aldehydes: 3CH3–CH2OH + K2Cr2O7 + 4H2SO4 = 3CH3–CHO + K2SO4 + Cr2(SO4)3 + 7H2O;
2) ang mga pangalawang alkohol ay na-oxidized sa mga ketone:

3) ang reaksyon ng oksihenasyon ay hindi tipikal para sa mga tertiary alcohol. Ang mga tertiary alcohol, sa mga molekula kung saan walang hydrogen atom sa carbon atom na naglalaman ng OH group, ay hindi nag-oxidize sa ilalim ng normal na mga kondisyon. Sa ilalim ng malupit na mga kondisyon (sa ilalim ng pagkilos ng malakas na oxidizing agent at sa mataas na temperatura), maaari silang ma-oxidized sa isang halo ng mababang molekular na timbang na mga carboxylic acid, i.e. ang pagkasira ng carbon skeleton ay nangyayari. Kapag ang methanol ay na-oxidized sa isang acidified na solusyon ng potassium permanganate o potassium dichromate, CO2 ay nabuo. Ang mga pangunahing alkohol sa panahon ng oksihenasyon, depende sa mga kondisyon ng reaksyon, ay maaaring bumuo ng hindi lamang aldehydes, kundi pati na rin ang mga acid. Halimbawa, ang oksihenasyon ng ethanol na may potassium dichromate sa malamig ay nagtatapos sa pagbuo ng acetic acid, at kapag pinainit, acetaldehyde:
3CH3–CH2OH + 2K2Cr2O7 + 8H2SO4 = 3CH3–COOH + 2K2SO4 + 2Cr2(SO4)3 + 11H2O,
3CH3–CH2OH + K2Cr2O7 + 4H2SO4
3CH3–CHO + K2SO4 + Cr2(SO4)3 + 7H2O
Naaalala namin ang impluwensya ng kapaligiran sa mga produkto ng mga reaksyon ng oksihenasyon ng alkohol, lalo na: ang isang mainit na neutral na solusyon ng KMnO4 ay nag-oxidize ng methanol sa potassium carbonate, at ang natitirang mga alkohol sa mga asin ng kaukulang mga carboxylic acid:

Oksihenasyon ng glycols
Ang 1,2-Glycols ay madaling mabulok sa ilalim ng banayad na mga kondisyon sa pamamagitan ng pagkilos ng periodic acid. Depende sa istraktura ng orihinal na glycol, ang mga produkto ng oksihenasyon ay maaaring aldehydes o ketones:
![]()
Kung tatlo o higit pang mga grupo ng OH ang nakagapos sa mga katabing carbon atoms, pagkatapos ay sa oksihenasyon na may periodic acid, ang gitna o gitnang mga atomo ay na-convert sa formic acid
Ang oksihenasyon ng glycols na may potassium permanganate sa isang acidic na kapaligiran ay katulad ng oxidative cleavage ng alkenes at humahantong din sa pagbuo ng mga acid o ketones, depende sa istraktura ng orihinal na glycol.
Aldehydes at ketones
Ang mga aldehydes ay mas madaling ma-oxidize kaysa sa mga alkohol sa kaukulang mga carboxylic acid hindi lamang sa ilalim ng impluwensya ng mga malakas na oxidant (air oxygen, acidified na solusyon ng KMnO4 at K2Cr2O7), ngunit din sa ilalim ng impluwensya ng mga mahina (ammonia solution ng silver oxide o tanso(II). ) hydroxide):
5CH3–CHO + 2KMnO4 + 3H2SO4 = 5CH3–COOH + 2MnSO4 + K2SO4 + 3H2O,
3CH3–CHO + K2Cr2O7 + 4H2SO4 = 3CH3–COOH + Cr2(SO4)3 + K2SO4 + 4H2O,
CH3–CHO + 2OH CH3–COONH4 + 2Ag + 3NH3 + H2O
Espesyal na atensyon!!! Ang oksihenasyon ng methanal na may ammonia solution ng silver oxide ay humahantong sa pagbuo ng ammonium carbonate kaysa sa formic acid: HCHO + 4OH = (NH4)2CO3 + 4Ag + 6NH3 + 2H2O.
Tulad ng nabanggit na, ang oksihenasyon ng isang organikong sangkap ay ang pagpapakilala ng oxygen sa komposisyon nito at (o) ang pag-aalis ng hydrogen. Ang pagbabawas ay ang kabaligtaran na proseso (pagpapakilala ng hydrogen at pag-aalis ng oxygen). Isinasaalang-alang ang komposisyon ng mga alkanes (СnH2n+2), maaari nating tapusin na hindi nila kayang lumahok sa mga reaksyon ng pagbabawas, ngunit maaaring lumahok sa mga reaksyon ng oksihenasyon.
Ang mga alkane ay mga compound na may mababang estado ng oksihenasyon ng carbon, at depende sa mga kondisyon ng reaksyon, maaari silang ma-oxidize upang bumuo ng iba't ibang mga compound.
Sa ordinaryong temperatura, ang mga alkane ay hindi tumutugon kahit na may malakas na mga ahente ng oxidizing (H2Cr2O7, KMnO4, atbp.). Kapag ipinasok sa isang bukas na apoy, nasusunog ang mga alkane. Sa kasong ito, sa labis na oxygen, ang kanilang kumpletong oksihenasyon ay nangyayari sa CO2, kung saan ang carbon ay may pinakamataas na estado ng oksihenasyon na +4, at tubig. Ang pagkasunog ng mga hydrocarbon ay humahantong sa pagkasira ng lahat ng C-C at C-H na mga bono at sinamahan ng paglabas ng isang malaking halaga ng init (exothermic reaction).
Karaniwang tinatanggap na ang mekanismo ng oksihenasyon ng mga alkane ay nagsasangkot ng isang radikal na proseso ng kadena, dahil ang oxygen mismo ay hindi maganda ang reaktibo upang ma-abstract ang isang hydrogen atom mula sa isang alkane, kailangan ng isang particle na magsisimula sa pagbuo ng isang alkyl radical, na kung saan; ay tutugon sa oxygen, na nagbibigay ng peroxy radical. Ang peroxy radical ay maaaring mag-abstract ng isang hydrogen atom mula sa isa pang alkane molecule upang bumuo ng isang alkyl hydroperoxide at radical.

Posibleng i-oxidize ang mga alkanes na may atmospheric oxygen sa 100-150 ° C sa pagkakaroon ng isang katalista - manganese acetate ang reaksyong ito ay ginagamit sa industriya. Ang oksihenasyon ay nangyayari kapag ang isang agos ng hangin ay tinatangay sa pamamagitan ng tinunaw na paraffin na naglalaman ng isang manganese salt.
kasi Bilang resulta ng reaksyon, ang isang halo ng mga acid ay nabuo, sila ay pinaghihiwalay mula sa unreacted paraffin sa pamamagitan ng dissolving sa may tubig alkali, at pagkatapos ay neutralized sa mineral acid.
Direkta sa industriya, ang pamamaraang ito ay ginagamit upang makakuha ng acetic acid mula sa n-butane:
Oksihenasyon ng mga alkenes
Ang mga reaksyon ng oksihenasyon ng mga alkenes ay nahahati sa dalawang grupo: 1) mga reaksyon kung saan napanatili ang carbon skeleton, 2) mga reaksyon ng oxidative na pagkasira ng carbon skeleton ng molekula sa double bond.
Mga reaksyon ng oksihenasyon ng mga alkenes na may pag-iingat ng balangkas ng carbon
1. Epoxidation (reaksyon ni Prilezhaev)
Ang mga acyclic at cyclic alkenes, kapag tumutugon sa mga peracid sa isang non-polar na kapaligiran, ay bumubuo ng mga epoxide (oxiranes).
Ang mga oxiranes ay maaari ding makuha sa pamamagitan ng oksihenasyon ng mga alkenes na may mga hydroperoxide sa pagkakaroon ng mga katalista na naglalaman ng molybdenum-, tungsten-, at vanadium:

Ang pinakasimpleng oxirane, ethylene oxide, ay ginawa sa industriya sa pamamagitan ng oksihenasyon ng ethylene na may oxygen sa pagkakaroon ng pilak o pilak na oksido bilang isang katalista.

2. anti-Hydroxylation (hydrolysis ng epoxides)
Ang acid (o alkaline) na hydrolysis ng mga epoxide ay humahantong sa pagbubukas ng oxide ring na may pagbuo ng mga transdiols.

Sa unang yugto, ang oxygen atom ng epoxide ay protonated upang bumuo ng isang cyclic oxonium cation, na nagbubukas bilang resulta ng nucleophilic attack ng molekula ng tubig.
Ang base-catalyzed epoxy ring opening ay humahantong din sa pagbuo ng mga trans-glycols.

3. syn-Hydroxylation
Ang isa sa mga pinakalumang paraan para sa oksihenasyon ng mga alkenes ay ang reaksyong Wagner (oksihenasyon na may potassium permanganate). Sa una, ang oksihenasyon ay gumagawa ng isang cyclic ester ng manganese acid, na nag-hydrolyze sa isang vicinal diol:

Bilang karagdagan sa reaksyon ng Wagner, mayroong isa pang paraan para sa syn-hydroxylation ng mga alkenes sa ilalim ng pagkilos ng osmium (VIII) oxide, na iminungkahi ni Krige. Kapag ang osmium tetroxide ay tumutugon sa isang alkene sa eter o dioxane, isang itim na precipitate ng cyclic osmic acid ester, osmate, ay nabuo. Gayunpaman, ang pagdaragdag ng OsO4 sa maramihang mga bono ay kapansin-pansing pinabilis sa pyridine. Ang nagreresultang black osmate precipitate ay madaling mabulok sa pamamagitan ng pagkilos ng isang may tubig na solusyon ng sodium hydrosulfite:

Ang potassium permanganate o osmium(VIII) oxide ay nag-oxidize ng alkene sa cis-1,2-diol.
Oxidative cleavage ng alkenes
Kasama sa oxidative cleavage ng alkenes ang mga reaksyon ng kanilang pakikipag-ugnayan sa potassium permanganate sa alkaline o sulfuric acid, pati na rin ang oksihenasyon na may solusyon ng chromium trioxide sa acetic acid o potassium dichromate at sulfuric acid. Ang huling resulta ng naturang mga pagbabago ay ang cleavage ng carbon skeleton sa site ng double bond at ang pagbuo ng mga carboxylic acid o ketones.
Ang mga monosubstituted alkenes na may terminal double bond ay nahati sa carboxylic acid at carbon dioxide:

Kung ang parehong mga carbon atom sa isang double bond ay naglalaman lamang ng isang alkyl group, kung gayon ang isang halo ng mga carboxylic acid ay nabuo:

Ngunit kung ang alkene tetrasubstituted sa double bond ay isang ketone:

Ang reaksyon ng ozonolysis ng mga alkenes ay may mas malaking kahalagahan sa paghahanda. Sa loob ng maraming dekada, ang reaksyong ito ay nagsilbing pangunahing pamamaraan para sa pagtukoy ng istruktura ng parent alkene. Ang reaksyong ito ay isinasagawa sa pamamagitan ng pagpasa ng isang kasalukuyang solusyon ng ozone sa oxygen, isang solusyon ng isang alkene sa methylene chloride o ethyl acetate sa -80 ... -100 ° C. Ang mekanismo ng reaksyong ito ay itinatag ni Krige:


Ang mga ozonides ay mga hindi matatag na compound na nabubulok nang paputok. Mayroong dalawang paraan ng pagkabulok ng ozonides - oxidative at reductive.
Sa panahon ng hydrolysis, ang mga ozonides ay nahahati sa mga carbonyl compound at hydrogen peroxide. Ang hydrogen peroxide ay nag-oxidize ng mga aldehydes sa mga carboxylic acid - ito ay oxidative decomposition:

Ang mas mahalaga ay ang reductive cleavage ng ozonides. Ang mga produkto ng ozonolysis ay aldehydes o ketones, depende sa istraktura ng panimulang alkene:
Bilang karagdagan sa mga pamamaraan sa itaas, mayroong isa pang pamamaraan na iminungkahi noong 1955 ni Lemieux:
Sa pamamaraang Lemieux, walang mga pamamaraang masinsinang paggawa para sa paghihiwalay ng manganese dioxide, dahil Ang dioxide at manganate ay muling na-oxidized ng periodate sa permanganate ion. Ito ay nagpapahintulot lamang sa catalytic na dami ng potassium permanganate na gagamitin.
4.5. Oksihenasyon ng mga alkenes
Maipapayo na hatiin ang mga reaksyon ng oksihenasyon ng mga alkenes sa dalawang malalaking grupo: mga reaksyon kung saan napanatili ang carbon skeleton at mga reaksyon ng oxidative na pagkasira ng carbon skeleton ng molekula sa double bond. Ang unang pangkat ng mga reaksyon ay kinabibilangan ng epoxidation, pati na rin ang hydroxylation, na humahantong sa pagbuo ng mga vicinal diols (glycols). Sa kaso ng cyclic alkenes, ang hydroxylation ay gumagawa ng vicinal kawalan ng ulirat- o cis-diols. Ang isa pang grupo ay kinabibilangan ng ozonolysis at kumpletong oksihenasyon na mga reaksyon ng alkenes, na humahantong sa pagbuo ng iba't ibang uri ng carbonyl compound at carboxylic acid.
4.5.a. Mga reaksyon ng oksihenasyon ng mga alkenes na may pag-iingat ng balangkas ng carbon
1. Epoxidation (reaksyon ni N.A. Prilezhaev, 1909)
Ang acyclic at cyclic alkenes, kapag tumutugon sa peracids (peracids) RCOOOH sa isang non-polar, walang malasakit na kapaligiran, ay bumubuo ng mga epoxide (oxiranes), kaya ang reaksyon mismo ay tinatawag na epoxidation reaction.
Ayon sa modernong katawagan IUPAC- isang singsing na may tatlong miyembro na may isang oxygen atom ay tinatawag na oxirane.
Ang epoxidation ng alkenes ay dapat isaalang-alang bilang isang kasabay, coordinated na proseso kung saan ang mga ionic intermediate tulad ng hydroxyl cation OH+ ay hindi lumalahok. Sa madaling salita, ang epoxidation ng alkenes ay isang proseso syn-attachment ng isang oxygen atom sa isang double bond na may kumpletong pangangalaga ng configuration ng mga substituents sa double bond.
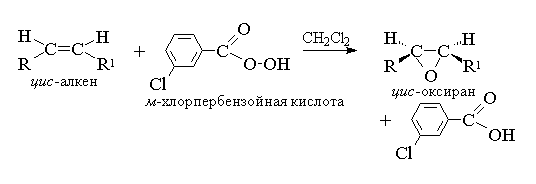

Ang isang mekanismo na katangian ng pinagsama-samang mga proseso ay iminungkahi para sa epoxidation.

Dahil ang pag-atake ng double bond ng oxygen atom ng peracid ay pantay na posibilidad sa magkabilang panig ng double bond plane, ang mga resultang oxiranes ay alinman meso-mga anyo, o pinaghalong mga enantiomer. Ang mga sumusunod na peracid ay ginagamit bilang mga epoxidizing agent: perbenzoic, m-chloroperbenzoic, monoperphthalic, peracetic, trifluoroperacetic at performic. Ang mga peracid ng aromatic series ay ginagamit sa anyo ng mga indibidwal na reagents, habang ang peracids ng aliphatic series - CH 3 CO 3 H, CF 3 CO 3 H at HCO 3 H ay hindi nakahiwalay sa indibidwal na anyo, ngunit ginagamit pagkatapos ng kanilang pagbuo sa ang interaksyon ng 30% o 90% hydrogen peroxide at ang kaukulang carboxylic acid. Perbenzoin at m-Ang chloroperbenzoic acid ay nakukuha sa pamamagitan ng oksihenasyon ng benzoic at m-chlorobenzoic acid na may 70% hydrogen peroxide sa isang solusyon ng methanesulfonic acid o mula sa acid chlorides ng mga acid na ito at hydrogen peroxide.


Ang monoperphthalic acid ay nakukuha sa katulad na paraan mula sa phthalic anhydride at 30% hydrogen peroxide.

Sa una, ang mga perbenzoic o monoperphthalic acid ay ginamit upang makakuha ng mga oxiranes (epoxides):

Sa kasalukuyan, ang pinakakaraniwang ginagamit para sa epoxidation ay m-chloroperbenzoic acid. Hindi tulad ng iba pang peracids, ito ay matatag sa loob ng mahabang panahon (hanggang 1 taon) at ganap na ligtas na hawakan. Mga ani ng oxiranes na nakuha mula sa oksihenasyon ng acyclic at cyclic alkenes m-chloroperbenzoic acid sa isang solusyon ng methylene chloride, chloroform o dioxane ay karaniwang mataas.



Ang mga peracid ay madalas na nabuo nang direkta sa isang reaksyong pinaghalong 90% hydrogen peroxide at carboxylic acid sa methylene chloride.


Ang mga alkenes na may double bond na pinagsama sa isang carbonyl group o iba pang acceptor substituent ay hindi aktibo at para sa kanilang oksihenasyon mas mainam na gumamit ng mas malakas na oxidizing agent, tulad ng trifluoroperacetic acid, na nakuha mula sa trifluoroacetic anhydride at 90% hydrogen peroxide sa methylene chloride. Ang pinakasimpleng oxirane, ethylene oxide, ay ginawa sa industriya sa pamamagitan ng oksihenasyon ng ethylene na may oxygen sa presensya ng pilak bilang isang katalista.

2. anti-Hydroxylation
Ang tatlong-member na singsing ng oxiranes ay madaling nagbubukas sa ilalim ng impluwensya ng isang malawak na iba't ibang mga nucleophilic reagents. Ang mga reaksyong ito ay tatalakayin nang detalyado sa seksyon ng acyclic at cyclic ethers. Dito lamang isasaalang-alang ang hydrolysis ng oxiranes. Ang hydrolysis ng oxiranes ay na-catalyzed ng parehong mga acid at base. Sa parehong mga kaso, ang mga vicinal diols, i.e. glycols, ay nabuo. Sa acid catalysis, sa unang yugto, ang oxirane oxygen atom ay protonated upang bumuo ng isang cyclic oxonium cation, na bubukas bilang resulta ng nucleophilic attack ng isang molekula ng tubig:

Ang pangunahing hakbang sa pagbubukas ng singsing, na tumutukoy sa rate ng buong proseso, ay ang nucleophilic attack ng tubig sa protonated form ng oxirane. Sa mekanikal, ang prosesong ito ay katulad ng pagbubukas ng isang bromonium ion sa pag-atake ng nucleophilic ng isang bromide ion o iba pang nucleophilic agent. Mula sa mga posisyong ito, ang resulta ng stereochemical ay ang pagbuo kawalan ng ulirat-glycols sa panahon ng pagkasira ng cyclic epoxides. Sa katunayan, sa panahon ng acid-catalyzed hydrolysis ng cyclohexene oxide o cyclopentene oxide, eksklusibo kawalan ng ulirat-1,2-diols.

Kaya, ang dalawang yugto na proseso ng alkene epoxidation na sinusundan ng acid hydrolysis ng epoxide sa pangkalahatan ay tumutugma sa reaksyon. anti-hydroxylation ng alkenes.
Parehong yugto anti-Ang hydroxylation ng mga alkenes ay maaaring pagsamahin kung ang alkene ay ginagamot ng may tubig na 30-70% hydrogen peroxide sa formic o trifluoroacetic acid. Ang parehong mga acid na ito ay sapat na malakas upang maging sanhi ng pagbubukas ng singsing ng oxirane.

Ang base-catalyzed na pagbubukas ng oxirane ring ay humahantong din sa pagbuo ng cyclic kawalan ng ulirat-glycols.

Samakatuwid, ang dalawang yugto na proseso ng epoxidation ng mga alkenes na sinusundan ng alkaline hydrolysis ng mga epoxide ay isa ring reaksyon anti-hydroxylation ng alkenes.
3. syn-Hydroxylation
Ang ilang mga asing-gamot at oksido ng mga metal na transisyon sa mas mataas na estado ng oksihenasyon ay mabisang mga reagents syn-hydroxylation ng double bond ng isang alkene, kapag ang parehong hydroxyl group ay idinagdag sa parehong bahagi ng double bond. Ang oksihenasyon ng mga alkenes na may potassium permanganate ay isa sa mga pinakalumang pamamaraan syn-Ang hydroxylation ng isang double bond ay patuloy na malawakang ginagamit sa kabila ng mga likas na limitasyon nito. Cis-1,2-cyclohexanediol ay unang nakuha ng V.V. Markovnikov noong 1878 sa pamamagitan ng hydroxylation ng cyclohexene na may tubig na solusyon ng potassium permanganate sa 0 0 C.

Ang pamamaraang ito ay kalaunan ay binuo sa mga gawa ng Russian scientist na si E.E. Wagner, samakatuwid syn-Ang hydroxylation ng mga alkenes sa ilalim ng pagkilos ng isang may tubig na solusyon ng potassium permanganate ay tinatawag na reaksyon ng Wagner. Ang potasa permanganeyt ay isang malakas na ahente ng pag-oxidizing na hindi lamang makakapag-hydroxylate ng double bond, kundi pati na rin sa pag-cleave sa resultang vicinal diol. Upang maiwasan ang karagdagang pagkasira ng glycols hangga't maaari, ang mga kondisyon ng reaksyon ay dapat na maingat na kontrolin. Ang mga ani ng glycols ay karaniwang mababa (30-60%). Ang pinakamahusay na mga resulta ay nakakamit sa pamamagitan ng hydroxylation ng mga alkenes sa isang bahagyang alkaline na kapaligiran (pH ~ 8 9) sa 0-5 0 C na may diluted na 1% aqueous solution ng KMnO 4.


Sa una, ang oksihenasyon ng mga alkenes na may potassium permanganate ay gumagawa ng isang cyclic ester ng manganese acid, na agad na na-hydrolyzed sa isang vicinal diol.

Ang cyclic ester ng manganese acid bilang isang intermediate ay hindi nakahiwalay, ngunit ang pagbuo nito ay sumusunod sa mga eksperimento na may label na 18 O potassium permanganate: ang parehong mga atomo ng oxygen sa glycol ay lumalabas na may label sa panahon ng oksihenasyon ng alkene KMn 18 O 4. Nangangahulugan ito na ang parehong mga atomo ng oxygen ay inilipat mula sa ahente ng oxidizing, at hindi mula sa solvent - tubig, na sumasang-ayon sa iminungkahing mekanismo.
Isa pang paraan syn-Ang hydroxylation ng alkenes sa ilalim ng impluwensya ng osmium (VIII) oxide OsO 4 ay iminungkahi ni R. Kriege noong 1936. Ang Osmium tetroxide ay isang walang kulay, pabagu-bago ng isip, mala-kristal na sangkap, lubos na natutunaw sa eter, dioxane, pyridine at iba pang mga organikong solvent. Kapag ang osmium tetroxide ay tumutugon sa mga alkenes sa eter o dioxane, isang itim na precipitate ng cyclic osmic acid ester ay nabuo - osmate, na madaling mabukod sa indibidwal na anyo. Ang pagdaragdag ng OsO 4 sa dobleng bono ay kapansin-pansing pinabilis sa solusyon sa pyridine. Ang agnas ng osmates sa vicinal glycols ay nakakamit sa pamamagitan ng pagkilos ng isang may tubig na solusyon ng sodium hydrosulfite o hydrogen sulfide.

Mga Produkto syn-hydroxylation ng alkenes sa paraang ito ay makabuluhang mas mataas kaysa kapag gumagamit ng permanganate bilang isang oxidizing agent. Ang isang mahalagang bentahe ng pamamaraang Krige ay ang kawalan ng mga produkto ng oxidative cleavage ng alkenes, na katangian ng permanganate oxidation.


Ang Osmium tetroxide ay isang napakamahal at mahirap makuhang reagent, at ito rin ay nakakalason. Samakatuwid, ang osmium (VIII) oxide ay ginagamit sa synthesis ng maliliit na dami ng mahirap maabot na mga sangkap upang makuha ang pinakamataas na ani ng diol. Para gawing simple syn-hydroxylation ng mga alkenes sa ilalim ng impluwensya ng OsO 4, isang pamamaraan ay binuo na nagpapahintulot sa paggamit ng mga catalytic na dami lamang ng reagent na ito. Ang hydroxylation ng mga alkenes ay isinasagawa gamit ang hydrogen peroxide sa pagkakaroon ng OsO 4, halimbawa:

Upang tapusin ang seksyong ito, ipinakita namin ang mga stereochemical na relasyon sa pagitan ng alkene cis- o kawalan ng ulirat-configuration at ang pagsasaayos ng nagreresultang vicinal diol, na maaaring cis- o kawalan ng ulirat-isomer, erythro- o tatlo-hugis, meso- o D,L-hugis depende sa mga substituent sa alkene:

Ang mga katulad na stereochemical na relasyon ay sinusunod sa iba pang mga reaksyon syn- o anti- karagdagan sa maramihang mga bono ng hydrogen, hydrogen halides, tubig, halogens, boron hydride at iba pang reagents.