Termodynamik för adsorptionsprocesser.
| Parameternamn | Menande |
| Artikelns ämne: | Termodynamik för adsorptionsprocesser. |
| Rubrik (tematisk kategori) | Utbildning |
Grundläggande definitioner och metoder för klassificering av adsorptionsprocesser.
Adsorption avser fenomen som uppstår på grund av en spontan minskning av ytenergi.
Adsorption– Processen för spontan reversibel eller irreversibel omfördelning av komponenterna i ett heterogent system mellan ytskiktet och volymen av den homogena fasen.
I flerkomponentsystem överförs företrädesvis den komponent som kraftigare minskar gränsytspänningen till ytskiktet. I enkomponentsystem, under bildandet av ytskiktet, sker en förändring i dess struktur (en viss orientering av atomer och molekyler, polarisation), kallad autoadsorption.
Den tätare fasen på vilken adsorptionsinteraktioner är lokaliserade kallas adsorbent. Ämnet som omfördelas mellan volymen av den homogena fasen och ytskiktet betecknas med termen '' adsorbatʼʼ.
I vissa fall är adsorptionsprocessen reversibel. I detta fall, under vissa förhållanden, kan en del av de adsorberade molekylerna som ett resultat av molekylära kinetiska fenomen röra sig från ytskiktet till bulkfasen. Den omvända processen för adsorption kallas desorption.
Metoder för att klassificera adsorptionsprocesser.
Klassificering av adsorptionsprocesser enligt tillståndet för aggregering av interagerande faser. Med hänsyn till beroendet av det aggregerade tillståndet för intilliggande faser, särskiljs följande typer av adsorptionsprocesser:
Adsorption av gaser på fasta adsorbenter;
Adsorption av lösta ämnen vid gränssnitten "fast-vätska" och "vätska-vätska".
Adsorption av ytaktiva ämnen vid vätske-gasgränsytan.
Klassificering av adsorptionsprocesser enligt mekanismen för interaktion mellan adsorbenten och adsorbatet. Adsorption kan betraktas som interaktionen mellan adsorbatmolekyler och adsorbentens aktiva centra. Enligt mekanismen för deras interaktion är följande typer av adsorption uppdelade:
1) fysisk (molekylär) adsorption– interaktionen mellan adsorbatets molekyler och adsorbenten utförs på grund av van der Waals-krafter, vätebindningar (utan kemiska reaktioner);
2) kemisk adsorption (kemisorption)– adsorbatmolekylernas fästning till adsorbentens aktiva centra sker som ett resultat av kemiska reaktioner av olika slag (med undantag för jonbytesreaktioner);
3) jonbytaradsorption (jonbyte) – omfördelning av adsorbatsubstansen mellan lösningen och den fasta fasen (jonbytaren) enligt jonbytesreaktionernas mekanism.
För att kvantitativt beskriva adsorptionsprocesser används två kvantiteter.
1) Absolut adsorption– kvantitet (mol) eller massa (kg) adsorbat per ytenhet eller massa av adsorbenten. Beteckning – A; dimension: mol/m2, mol/kg, kg/m2, kg/kᴦ.
2) Gibbs (överskott) adsorption– överskott av adsorbatämne i ett ytskikt av en viss tjocklek jämfört med dess mängd i volymen av den homogena fasen, per ytenhet eller massa av adsorbenten. Beteckning – G; dimension: mol/m 2, mol/kᴦ.
Förhållandet mellan absolut och överskottsadsorption kan illustreras med hjälp av ekvationen:
Г = А – с * h (3,1)
där c är jämviktskoncentrationen av ämnet i fasens volym, mol/m3;
h är tjockleken på ytskiktet, som konventionellt antas vara 10 -9 m.
I flerkomponents heterogena system, när en eller annan komponent omfördelas mellan volymen av den homogena fasen och ytskiktet, är ekvationen för ytans överskott av inre energi giltig:
U = T * S + s * s + Sm i * n i (3,2)
Genom att reducera alla termer i ekvationen till enhetsarea för interfasytan får vi:
U s = T * S s + s + Sm i * Г i (3.3)
där Г i = n i/s är överskottet av den i:te komponenten i ytskiktet, det vill säga Gibbs adsorption.
För ett enkomponentsystem kommer ekvation (3.3) att ha formen:
G s = s + m * G (3,4)
där G s = U s - T * S s – Gibbs energi av ytan eller arbetet med att skapa en enhetsytarea;
m * G – packning av det adsorberade ämnets substans i ytskiktet.
Baserat på ekvation (3.4) kan vi dra slutsatsen att under adsorption består arbetet med att skapa en interfasyta av arbetet med ytbildning (brytning av kohesiva bindningar i volymen av adsorbatfasen) och packning av ämnet i ytskiktet.
I ett tillstånd av dynamisk jämvikt mellan adsorbenten och adsorbatet, förändringen i Gibbs-energin i det heterogena systemet ΔG = 0, beskrivs termodynamiken för adsorptionsprocessen med ekvationen som kallas Gibbs fundamentala adsorptionsekvation:
Ds = SГ i * dm i (3,5)
Denna ekvation är universell, eftersom den är giltig för alla typer av adsorptionsprocesser
Specialfall av Gibbs adsorptionsekvation.
1) Adsorption från lösningar.
För den kemiska potentialen för den i:te komponenten i systemet under adsorption vid gränssnitten "vätska - fast adsorbent" och "vätska - gas" är följande ekvationer giltiga:
m i = m i 0 + R*T*ln a i (3,6)
dm i = R*T* d ln a i (3,7)
där m i 0 är den kemiska potentialen för den i:te komponenten i systemet under standardförhållanden;
a i är aktiviteten för den i:te komponenten i systemet under standardförhållanden.
Baserat på detta tar Gibbs adsorptionsekvation formen:
Г i = - a i / R*T * (ds / da i) (3.8)
För lösningar av icke-elektrolyter tar vi a i = c i, då:
Г i = - с / R*T * (ds / dс) (3.9)
För elektrolytlösningar:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
där с ± är den genomsnittliga jonkoncentrationen av lösningen;
n är den stökiometriska koefficienten.
2) Adsorption av ämnen från gasfasen.
I enlighet med Mendeleev-Clayperons ekvation:
Р = с * R*T (3,11)
I detta avseende skrivs Gibbs ekvation för adsorption av gaser på fasta adsorbenter i följande form:
Г i = - Р / R*T * (ds / dР) (3.12)
I praktiken tillåter Gibbs adsorptionsekvation, baserat på ytspänningsmätningar vid olika värden på vätskekoncentration eller jämviktsgastryck, att beräkna mängden adsorption av ämnen i gränsskiktet för vilken ytspänningen bestäms.
Termodynamik för adsorptionsprocesser. - koncept och typer. Klassificering och funktioner i kategorin "Termodynamik för adsorptionsprocesser." 2017, 2018.
Aktuell sida: 6 (boken har totalt 19 sidor) [tillgänglig läsning: 13 sidor]
Font:
100% +
34. Typ av adsorptionskrafter
Interaktionen mellan adsorbentmolekyler med ytan av adsorbenten under den sk. fysisk adsorption kan bero på olika orsaker. Då kan potentialen som bestämmer interaktionen mellan en adsorbentmolekyl och en atom i en opolär adsorbent uttryckas på följande sätt:
θ = −Cr 6 +Br 12 ,
där r är avståndet mellan partiklarnas centra; C – dispersionsattraktionskonstant; B är en konstant som kännetecknar energin hos frånstötande krafter.
Det är helt uppenbart att på relativt avlägsna avstånd bör attraktionskrafterna råda, och på nära avstånd bör avstötningskrafterna råda. Även på vissa avstånd måste dessa krafter vara lika, vilket kommer att motsvara ett minimum av fri energi. Men det är viktigt att notera att under adsorption verkar dispersionskrafter samtidigt mellan varje opolär partikel.
Eftersom partiklars interaktionsenergi snabbt kan minska med avståndet, räcker det för att bestämma potentialen för adsorptionskrafter att utföra summering på de närmaste atomerna i adsorbenten. Det är viktigt att under adsorptionen av komplexa opolära molekyler kan den potentiella energin ungefärligen beräknas som summan av alla potentiella adsorptionsenergier för molekylenheterna.
Om adsorbenten består av joner, kan verkan av de redan kända dispersionskrafterna kompletteras med verkan av de induktiva attraktionskrafterna av dipoler som induceras i adsorbentens molekyler av det elektriska fältet, vilket i sin tur är skapas av jonerna i det adsorberande gittret.
Med en sådan växelverkan kan andelen induktiva krafter i adsorptionsväxelverkan vara proportionell mot adsorbentmolekylens polariserbarhet och kvadraten på fältstyrkan på denna adsorbentyta.
Om adsorptionen av polära molekyler av adsorbenten sker på en polär adsorbent, så polariserar dipolerna i detta fall adsorbentens atomer, d.v.s. de verkar inducera elektriska moment i dem. På grund av denna påverkan läggs den induktiva interaktionen till dispersionsinteraktionen.
Den induktiva interaktionen i sig är vanligtvis liten och kan, beroende på adsorbentmolekylens dipol och adsorbentens polariserbarhet, nå stora värden. Om molekyler adsorberas på en adsorbent som har joner eller dipoler på ytan, den s.k. växelverkan mellan joner eller dipoler hos adsorbenten med det elektrostatiska fältet hos själva adsorbenten.
I detta fall kan adsorbentens molekyler till och med vara orienterade i adsorbentens fält och orienterande Coulomb-interaktion inträffar. Det händer vanligtvis att energierna för induktiva och orienterande interaktioner är mindre än energin för dispersiva interaktioner, och därför är det accepterat att energin för intermolekylär attraktion bestäms av energin för dispersiv attraktion.
Adsorption kan också orsakas av bildandet av en vätebindning. En bindning av denna typ kan uppstå under adsorption på adsorbenter som innehåller hydroxylgrupper av molekyler som vatten, alkoholer, ammoniak och aminer på ytan. När en vätebindning bildas kan interaktionsenergin mellan adsorbenten och adsorbenten vara ganska stor, och värmen som frigörs under sådan adsorption är betydligt större än adsorptionsvärmen för ämnen som liknar form och storlek på molekyler. , men bildar inte en vätebindning.
Det är viktigt att notera att genom att känna till den termodynamiska beskrivningen av ytskiktet vid gränssnittet adsorbent-adsorbent, dess struktur, arten av olika typer av krafter och dynamiken i processen, kan man gå vidare till studiet av mer komplex adsorption processer.
35. Adsorption som spontan koncentration vid gränsytan av ämnen som minskar gränsytspänningen
Ytaktiva ämnen delas in i två stora grupper: aktiv och inaktivämnen.
Ytaktiva ämnen kan ansamlas i ytskiktet och positiv adsorption uppstår G > 0.
Dessa typer av ämnen måste ha en ytspänning, som i sin tur måste vara mindre än lösningsmedlets ytspänning, annars blir ansamlingen av ämnet i ytskiktet ogynnsam och måste ha relativt låg löslighet. Med tillräckligt god löslighet tenderar ytaktiva molekyler att röra sig från ytan till lösningens djup. Följaktligen kommer ytaktiva ämnen företrädesvis att tryckas ut ur huvuddelen av vätskan till ytan.
Men med ackumuleringen av ämnen vid lösningens gräns i molekylerna av dessa ämnen, som svagt interagerar med varandra, kommer den intermolekylära interaktionen i ytskiktet att minska, och ytspänningen kommer att falla.
Ytaktiva ämnen i förhållande till vattenskiktet finns många typer av organiska föreningar, fettsyror med en ganska stor kolväteradikal, salter av dessa syror (tvålar), sulfonsyror och deras salter, samt olika typer av alkoholer och aminer. En karakteristisk egenskap hos de flesta molekyler är deras difilicitet: molekylen består av två delar av en polär grupp och en opolär kolväteradikal. En polär grupp som har ett betydande dipolmoment och är mycket hydratiserande kan bestämma det ytaktiva medlets affinitet för den vattenhaltiga miljön. Men kolväteradikalen är orsaken som minskar lösligheten av dessa föreningar.
Yt-inaktiva ytaktiva ämnen- dessa typer av ämnen tenderar att lämna vätskans yta i sin volym, vilket resulterar i den sk. negativ adsorption G < 0. Поверностно-инактивные вещества также обладают значительным поверхностным натяжением, значительно большим, чем натяжение у растворителя (иначе эти вещества способны самопроизвольно накапливаться в поверхностном слое), также обладают высокой растворимостью, что способствует их стремлению уйти с поверхности жидкости в объем. Взаимодействие между молекулами поверхностно-инактивного вещества и растворителя всегда больше, чем взаимодействие между самими молекулами растворителя, поэтому они и стремятся перейти в объем раствора. Ytanaktiva ämnen I förhållande till vatten finns det många oorganiska elektrolyter: syror, alkalier, salter. Ytaktiva molekyler har ingen hydrofob del och kan sönderfalla i vatten till mycket hydratiserande joner.
Exempel Ytaktiva ämnen är också några organiska föreningar där den opolära delen av molekylen är frånvarande eller mycket liten. Dessa ämnen inkluderar myrsyra och aminoättiksyror.
I icke-vattenhaltiga lösningsmedel kan oorganiska elektrolyter också öka ytspänningen, beroende på lösningsmedlet.
Till exempel När natriumjodid införs i metanol ökar ytspänningen kraftigt, för etanol är ytspänningen ungefär 2 gånger högre. Ämnes ytaktivitet kan bero inte bara på ämnets natur utan också på lösningsmedlets egenskaper. Om något lösningsmedel har en hög ytspänning kan det lösta ämnet uppvisa betydande ytaktivitet.
36. Adsorptionsteorier
Låt oss överväga de vanligaste adsorptionsteorierna som beskriver vissa typer av adsorption vid gränssnittet "fast gas" eller "fast lösning".
Teorin om monomolekylär adsorption av I. Langmuir.
1. Adsorptionen är lokaliserad och orsakas av krafter nära kemiska.
2. Adsorption sker endast på aktiva centra - utsprång eller fördjupningar på ytan av adsorbenten, kännetecknad av närvaron av fria valenser. De aktiva centren anses vara oberoende och identiska.
3. Varje aktivt centrum kan interagera med endast en adsorbatmolekyl; Endast ett lager av adsorberade molekyler kan bildas på ytan.
4. Adsorptionsprocessen är reversibel och i jämvikt; den adsorberade molekylen hålls kvar av det aktiva stället under en tid, varefter den desorberas; Efter en tid etableras dynamisk jämvikt.
Maximalt möjligt adsorptionsvärde G o uppnås förutsatt att alla aktiva centra är upptagna av adsorbatmolekyler. Monomolekylär adsorptionsisoterm ekvation som relaterar storleken på adsorptionen G med adsorbatkoncentration MED, har formen:
Var b– ett konstant värde för ett givet "adsorbent - adsorbat"-par (förhållandet mellan hastighetskonstanterna för desorption och adsorption), numeriskt lika med adsorbatkoncentrationen vid vilken hälften av de aktiva centran är upptagna.

Langmuirs adsorptionsisotermgraf visas i figur 2. Konstanten b bestämma grafiskt genom att dra en tangent till adsorptionsisotermen vid punkten MED= 0. Vid beskrivning av gasadsorptionsprocessen i ekvationen kan koncentrationen ersättas med ett proportionellt värde på partialtrycket. Teori om monomolekylär adsorption I. Langmuir tillämplig för att beskriva processerna för adsorption av gaser och lösta ämnen vid låga tryck (koncentrationer) av adsorbatet.
Polyanis teori om polymolekylär adsorption beskriver s-formade adsorptionsisotermer, vars form indikerar möjlig interaktion mellan adsorberade molekyler och adsorbatet.
1. Adsorption orsakas av fysiska krafter.
2. Adsorbentens yta är homogen, det finns inga aktiva centra; adsorptionskrafter bildar ett kontinuerligt kraftfält nära ytan av adsorbenten.
3. Adsorptionskrafter verkar på ett avstånd som är större än adsorbatmolekylens storlek, d.v.s. det finns en viss adsorptionsvolym vid ytan av adsorbenten, som fylls med adsorbatmolekyler under adsorptionen.
4. Attraktionen av en adsorbatmolekyl av adsorbentens yta beror inte på närvaron av andra molekyler i adsorptionsvolymen, vilket resulterar i att polymolekylär adsorption är möjlig.
5. Adsorptionskrafter beror inte på temperaturen, och därför ändras inte adsorptionsvolymen med en förändring i temperaturen.
Freundlichs ekvation. Ytan på adsorbenten är heterogen, interaktion sker mellan de adsorberade partiklarna och de aktiva centran är inte helt oberoende av varandra. G. Freundlich föreslog att antalet mol adsorberad gas eller löst ämne per massenhet av adsorbenten (den så kallade specifika adsorptionen X/m), måste vara proportionell mot jämviktstrycket (för gas) eller jämviktskoncentrationen (för ämnen adsorberade från lösning) av adsorbenten, höjt till en viss effekt, som alltid är mindre än ett:
x / m = aP n; x / m = aC n.
Exponenter n och proportionalitetsfaktor A bestäms experimentellt.
37. Termodynamik för adsorptionsprocessen. Gibbs adsorptionsekvation
För att studera fenomenet med adsorption vid "lösning-gas"-gränssnittet är det nödvändigt att upprätta ett samband mellan överskottet av det adsorberade ämnet i lagret på ytan ( G), tensidkoncentration i lösning ( Med) och ytspänning ( σ ) vid fasgränsen "lösning - gas". Det är mer ändamålsenligt att betrakta fenomenen ur en termodynamisk synvinkel och relatera adsorptionen av ett löst ämne till en förändring i ytans fria energi eller dess ytspänning. Denna koppling skapades W. Gibbs V 1876, som namngavs "Gibbs adsorptionsekvation":
G = – Med / RT x dσ/dc.
Du kan fortfarande föreställa dig Gibbs ekvation, baserad på termodynamik, med användning av isobarisk-isotermisk potential G kemiska potentialer μ 1 Och μ2, och även använda n 1 Och n 2 antal mol komponenter. Efter att ha analyserat det med hänsyn till entropi S, volym V och tryck P, kan vi skriva följande ekvation:
dG=– SDT+VdP+σds+ μ 1 dn 1+ μ 2 dn 2.
Låt oss likställa det med noll, och med hänsyn till konstant temperatur och tryck, förenklas det till en ekvation av formen:
SD σ + n 1 d μ 1 + n 2 d μ 1 = 0.
Med hänsyn till det faktum att för utspädda lösningar uttrycks den kemiska potentialen för den andra komponenten enligt följande:
μ 2 = μ 2 0 +RT ln c,
och givet att temperaturen är konstant
dμ 2 =RTdnc,
ersätta denna ekvation med
![]()
vi får den önskade Gibbs adsorptionsekvation. Utifrån ekvationen kan man se att om ytspänning σ ökar med koncentrationen Med, då är koncentrationen av det lösta ämnet på ytskiktet mindre än i lösningens bulk (så kallad negativ adsorption), och om ytspänningen σ minskar med ökande koncentration Med, då är koncentrationen i skiktet större än i volymen (positiv adsorption), och slutligen, om σ inte beror på Med, då är koncentrationen av ämnet i skiktet på ytan och i volymen densamma. Gibbs ekvation härleddes med användning av termodynamik. I praktiken är det svårt att verifiera denna ekvation, vilket beror på svårigheten att bestämma koncentrationen av det lösta ämnet i skiktytan. Erfaret sätt B. McBen fann att ett mycket tunt lager vätska skars av från lösningens yta med hjälp av anordningen. Ytterligare bestämning av alla parametrar i Gibbs ekvation visade att de experimentellt hittade adsorptionsvärdena, inom det experimentella felet, sammanföll med värdena som beräknades med Gibbs ekvation. På grund av homogeniteten och jämnheten hos ytan av någon vätska är de vanliga koncepten för aktiva centra helt otillämpliga när man studerar adsorption på dess yta. Vid en kritisk temperatur försvinner skillnaden mellan de intilliggande faserna och ytspänningen blir som regel lika med noll. Adsorption av gaser och ångor har en så bred praktisk tillämpning att man i litteraturen, särskilt i teknisk litteratur, kan hitta detta koncept, som endast tillämpas i förhållande till processer på ytan av fasta ämnen.
Detta koncept, liksom de mest allmänna adsorptionslagarna, liksom Gibbs ekvation som betraktas, är tillämpligt på alla fasgränser. Med hjälp av Gibbs ekvation och alla bestämmelser som följer av den, efter att ha bestämt värdet på Γ, är det möjligt att konstruera en adsorptionsisoterm.
38. Egenskaper för adsorption på mikroporösa material. Polyanys potentiella teori. Adsorptionspotential
Polyanis teoriöverväger icke-lokaliserad fysisk adsorption, som är direkt orsakad av van der Waals-krafter mellan adsorbenten och adsorbatet (detta kan betraktas som den första positionen). Den andra positionen i denna teori är idén om ett kraft- (eller potentiellt) fält för adsorbenten, som sträcker sig över ett avsevärt avstånd från ytan; adsorptionsskiktet som uppträder i detta fält är polymolekylärt. Om vi betraktar adsorptionen av gaser, minskar tätheten av detta lager längs en viss normal från ytan. Om vi överväger ångadsorption, bildas ett vätskeskikt av en viss tjocklek på ytan. Fältet i Polyanis teori betraktas som en serie ekvipotentiella ytor, varje yta motsvarar ett visst potentiellt värde ε , och varje efterföljande yta kommer att vara mindre än den föregående. Varje sådan yta i rymden skär ut lager av en viss volym, betecknad som v i. Uppgiften för Polyanyis teori är att hitta övergången från de vanliga koordinaterna för isotermen ( x, sid) till fältparametrar εi Och v i, med ytterligare etablering av sambandet mellan dessa grundläggande parametrar. Den första delen av problemet, som Polyany lade upp, är ganska komplex och kan i många fall inte ha definitiva lösningar, men för fallet med ångadsorption löses denna del av problemet i en första approximation mycket enkelt. För ett vätskeadsorptionsskikt kommer den fyllda delen av volymen att vara lika med:
v i = x(M/d),
Var d– ämnets densitet i flytande tillstånd.
I sin teori introducerar M. Polyany en annan bestämmelse om frånvaron av den sk. fältscreening under adsorption, värde ε i denna teori är rymden ett konstant värde (något liknande gravitationspotentialen) oavsett om vissa adsorbatmolekyler existerar mellan en given punkt och en fast yta eller om allt utrymme är fritt. Polyani introducerar konceptet adsorptionspotential ε , som representerar det isotermiska arbetet med kompression av ånga när den överförs från jämviktstryck R i bulkfasen bort från ytan in i området för ytskiktet med mättat ångtryck p 0 då kommer uttrycket för att bestämma potentialen se ut så här:
ε = RT ln R 0 / R.
Med hjälp av denna ekvation kan du gå från koordinater x, p till koordinater ε Och v och få en kurva, som kallas "karakteristisk". Polyani upptäckte i sina experiment att sådana kurvor, konstruerade från experimentella data från de erhållna isotermerna, har följande egenskap: de är invarianta med avseende på T, eller, med andra ord, alla kurvor av denna typ kan passa på en kurva ε −ε .
M. Polyany accepterade denna position som ett postulat, dvs.
Denna Polyani-egenskap är av stor praktisk betydelse; den kan konstruera en familj av isotermer från en experimentell adsorptionsisoterm.
Polanyis teori ger inte ett analytiskt uttryck för isotermen eller potential-volymfunktionen, men den tillåter en att beräkna koordinaten för en given temperatur om minst en isoterm är känd. Detta resultat är mycket viktigt för tekniska beräkningar, eftersom för liknande gaser på samma adsorbent kan adsorptionskurvorna ligga nära varandra och kan kombineras i många fall.
39. Karakteristisk adsorptionskurva. Temperaturinvarians och affinitet för karakteristiska kurvor
Kraftfältet som uppstår vid ytan av adsorbenten kan på många sätt likna gravitationsfältet. I adsorptionsfältet kan man tänka sig potentiella ytor, det vill säga ytor som kännetecknas av samma adsorptionspotential. Under begreppet adsorptionspotential θ ska inte förstås som något annat än det arbete som utförs mot adsorptionskrafter när man flyttar 1 mol adsorbent från en viss punkt i fältet till en viss gasfas. Den maximala adsorptionspotentialen kommer att finnas vid gränsen "adsorbent – adsorptionsvolym". Men vid gränsen "volym - gasfas" (det är där verkan av adsorptionskrafter slutar), bör adsorptionspotentialen vara lika med noll. Förändringen i adsorptionspotential med en förändring i adsorptionsvolymen kan representeras i form av kurvor. Detta gjordes för första gången av M. Polyani. Dessa typer av kurvor är inte beroende av temperatur och kan vara karakteristiska för varje specifik adsorbent, dessa typer av kurvor brukar kallas karakteristiska adsorptionskurvor. Teorin för polymolekylär adsorption antar att gastillståndsekvationen är tillämplig för adsorptionsvolymen. Följaktligen liknar de isotermer som kännetecknar beroendet av densiteten hos adsorbenten på volymen för olika temperaturer isotermerna för tryckets beroende av volymen. Vid låga temperaturer kan ytadsorptionskrafter göra att ånga kondenserar till en vätska med en viss densitet. Vid lägre temperaturer än kritiskt, under kondensation, kommer hela adsorptionsvolymen att fyllas med vätska. I detta fall kommer adsorptionskurvan att löpa nästan parallellt med abskissaxeln, vilket är förknippat med vätskans låga kompressibilitet. Sedan sjunker adsorptionskurvan vid gränsen för "volym - gasfas" kraftigt, och följaktligen når densiteten hos adsorbenten en viss densitet av gasfasen. Vid temperaturer högre än den kritiska temperaturen kan adsorptiven bete sig som en idealgas, och grafen kommer att uttryckas som en ideal gasisoterm, förutsatt att pV = RT. Under sådana förhållanden kommer den adsorberade gasen att ha en maximal densitet vid själva ytan av adsorbenten och en minimal densitet i omedelbar närhet av gasfasen. Dessutom är det i detta fall viktigt att notera att densiteten hos adsorbenten i adsorptionsskiktet ingenstans når densiteten hos själva vätskan. Och om temperaturen är mycket nära kritisk, kommer densitetens beroende av volymen att uttryckas av en kurva nära isotermen, vilket beskrivs van der Waals ekvation. I denna situation kommer en del av den adsorberade substansen att vara i den adsorberade volymen i flytande tillstånd, och en del av den adsorberade substansen kommer att vara i ett gasformigt tillstånd. Då kommer kurvan att minska kraftigast i det avsnitt som motsvarar övergången från vätska till gas. Om du konstruerar en karakteristisk kurva från den experimentella adsorptionsisotermen för en av adsorptiva, och känner till motsvarande affinitetskoefficienter för något annat adsorptiv, kan du hitta adsorptionsisotermen och konstruera den för en annan adsorptiv. Potentialteorin för adsorption gör det möjligt att beräkna olika adsorptionsisotermer för olika ångor på samma adsorbent, med hjälp av en karakteristisk kurva som erhålls från adsorptionsisotermen för en ånga, eftersom förhållandet mellan adsorptionspotentialen inte beror på adsorptionsvolymerna .
Affinitet(från latin affinis - "relaterad") - affinitetskromatografi. Metoden för att rena och separera proteiner är baserad på deras selektiva interaktion med en ligand kovalent bunden till en inert bärare (affinitetskromatografi). Att mäta affiniteten hos ett toxiskt ämne för en receptor är i huvudsak en experimentell studie av förhållandet mellan mängden av ett ämne som tillsätts till inkubationsmediet och mängden toxiskt-receptorkomplex som bildas som ett resultat av interaktion.
Adsorption sker i gränssnittet. Därför är det rimligt att betrakta den termodynamiska beskrivningen av ytfenomen som ett specialfall av termodynamiken i heterogena system.
Ris. 3.4. Gibbs adsorption: 1- tvåfas jämförelsesystem, 2- verkligt tvåfassystem med en olikformig region
I termodynamiken för heterogena system används det additivitetsprincipen vilket är följande: alla extensiva egenskaper hos ett heterogent system är lika med summan av motsvarande extensiva egenskaper som faserna skulle ha haft innan de kom i kontakt. Låt oss beteckna faserna med α och β (fig. 4). För ett idealiskt system, så att egenskaperna för faserna nära gränssnittet sammanfaller med deras bulkegenskaper, gäller följande relationer för den inre energin U, volym V, massa (antal mol) n, entropi S efter upprättande av jämvikt i ett heterogent system:
U = U α + U β, V = V α + V β, n = n α + n β, S = S α + S β
Detta förutsätter att temperaturen och trycket i båda faserna är desamma.
För verkliga heterogena system ger övergångsregionen vid gränsen av två faser ytterligare ett bidrag till systemets omfattande egenskaper. Om ytfenomen uppstår bör man ta hänsyn till skillnaden mellan de omfattande egenskaperna hos ett verkligt heterogent system och de omfattande egenskaperna hos ett modellsystem där ytfenomen saknas. Ett sådant system kallas ett jämförelsesystem. Jämförelsesystemet har samma intensiva parametrar (T, P, Ci ...) och samma volym V som det verkliga systemet (Fig. 4).
Ur termodynamisk synvinkel förstås adsorptionsvärdet G som den överskottsmängd av ämne n s, uttryckt i mol eller gram, som ett verkligt heterogent system har i jämförelse med referenssystemet, relaterat till gränsytan eller till ytan. av adsorbenten A. Det antas att jämförelsesystemet har samma intensiva parametrar (T, P, Ci) och samma volym (V = V α + V β) som det verkliga systemet (fig. 4) .
Г = (n - n a - n p)/A = n s/A 3.11
Överskott av termodynamiska funktioner i övergångsområdet för ett reellt system (vi betecknar dem med index s) kan skrivas som
U s = U - U α - U β , n s = n - n α - n β , S s = S - S α - S β etc.
Experimentella mätningar av adsorption ger alltid adsorption exakt som ett överskott av komponenten i det verkliga systemet jämfört med det valda referenssystemet. Till exempel, vid adsorption av gas på en fast adsorbent eller vid adsorption av komponenter på en fast fas, för att hitta adsorptionsvärden, bestäm förändringen i de initiala koncentrationerna av adsorbatet efter kontakten av faserna α och β
n i s = V(Ci o - C i),
Var C i o– initial koncentration av den i:te komponenten, C i– Koncentration av den i:te komponenten efter upprättande av jämvikt mellan de kontaktande faserna. Man tror att volymen Vändras inte. Däremot koncentrationen i komponenten C i Experimentellt erhållen bestäms i volym V' ovanför fasgränsytan utan att ta hänsyn till volymen av den inhomogena regionen av övergångsskiktet V a vid gränssnittet där koncentrationen är Cia. På grund av att det finns en olikformig region i ett verkligt system kan systemets totala volym representeras som V = V' + V α. All kvantitet i-te komponenten C i o kommer att distribueras mellan dessa två volymer:
V C i o = V’ C i + V α C i α ,
och antalet mol av komponenten i, adsorberad på gränssnittet, kommer att vara lika med
n i s = (V’C i + V α C i α) – (V’ + V α)C i = V α (Ci α – C i) 3.12
De där. experimentellt bestämd adsorption är överskottet av den i:te komponenten i volymen V α jämfört med mängden av denna komponent i samma volym långt från fasgränsytan. Denna typ av adsorption kallas Gibbs adsorption. .
V aCi a kallas fullständigt innehåll jag- komponenten i adsorptionsskiktet. I området med mycket låga koncentrationer C i i volym V'ändring V aCi ekvation (3.2) kan försummas och det uppmätta värdet kan beaktas V aCi a fullständigt innehåll jag- komponenten i adsorptionsskiktet, till exempel under gasadsorption på en fast adsorbent vid låga tryck.
Adsorption som en spontan koncentration av molekyler på en yta åtföljs av en minskning av systemets entropi. Eftersom kriteriet för processens spontanitet är
∆H - T · ∆S = ∆G< 0,
då är adsorption endast möjlig vid ∆H< 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. När temperaturen ökar skiftar jämvikten mot den endotermiska processen, dvs desorption.
Adsorption på fast yta
1. Monomolekylär adsorption.
Enligt Langmuirs teori interagerar adsorberande molekyler med ytan av adsorbenten och bildar slutligen ett monomolekylärt skikt. I detta fall, graden av fyllning () av ytan med det adsorberade ämnet under adsorption från gasfasen
från vätska
där K är jämviktskonstanten (adsorptionskonstanten);
p är partialtrycket för den adsorberade gasen;
c är koncentrationen av det adsorberade ämnet.
Beroendet av β på p (eller c) presenteras av grafen (adsorptionsisoterm, T = const) i fig. 1.3.

Ris. 1.3. Ytfyllnadsgrad med adsorberat ämne
Vid låga koncentrationer och partialtryck är adsorptionen proportionell mot koncentrationen eller partialtrycket:
R<< 1, β ≈ К· r ilis<< 1, β ≈ К· si e. den initiala sektionen av isotermen är ungefär linjär och tan α = K (tg α bestäms av kurvans lutning vid p (eller c) → 0: eller ).
Om är antalet mol adsorberat ämne per 1 g adsorbent; - det högsta möjliga antalet mol adsorberat ämne per 1 g adsorbent ("monolagerkapacitet"), sedan
Ersätter β i ekvation (1.3) (för fallet med adsorption från gasfasen, koncentrationen Med i ekvationer bör ersättas med tryck R), vi får:
 (1.6)
(1.6)
Eftersom och K i ett givet adsorbent-adsorbentpar är konstanter (vid T=const), då kan man genom beroende hitta TILL(Fig. 1.4).

Ris. 1.4. Grafisk lösning av adsorptionsekvationen
erhålls genom att extrapolera det experimentella linjära beroendet till () = 0; och sedan , då .
Värdet kan användas för att bestämma adsorbentens specifika yta UD (i m 2 per 1 g adsorbent), om arean ω som upptas på ytan av en molekyl av adsorbenten är känd (bestäms utifrån molekylens storlek):
UD = · ω · Na, (1,7)
där Na är Avogadros tal (Na = 6,02 10 23).
I sin tur kan det kända värdet på UD användas för att beräkna ω för vilket ämne som helst baserat på dess adsorption på en given adsorbent.
2. Polymolekylär adsorption.
Ekvation (1.5) beskriver en kurva med mättnad, d.v.s. på
p (eller c) → ∞ tenderar mot gränsvärdet lika med (Fig. 1.5,a).

Fig.1.5. Adsorptionsisotermer:
a – adsorption med mättnad; b – polymolekylär adsorption
Men i vissa fall ser adsorptionsisotermer ut som de som visas i fig. 1,5, b, dvs. når inte gränsen ens vid hög p (eller c).
Beroenden av den typ som visas i fig. 1,5,b motsvarar polymolekylär adsorption. Som regel är sådana isotermer karakteristiska för ämnen med starka intermolekylära interaktioner (till exempel vatten). När adsorptionscentra på ytan av adsorbenten är upptagna (det monomolekylära lagret är mättat) sker "landningen" av nästa adsorbatmolekyler på grund av intermolekylära interaktioner med redan adsorberade molekyler (Fig. 1.6). Värmen av sådan adsorption är nära i absolut värde, men motsatt i tecken till värmen från avdunstningen av motsvarande vätska (tänk på varför).

Fig.1.6. Adsorptionsschema:
a - monomolekylär adsorption; b - polymolekylär adsorption
När vi kommer närmare R till det mättade ångtrycket hos det adsorberade ämnet börjar det kondensera på ytan av adsorbenten, som ett resultat av det växer det snabbt med ökande R.
Vid interaktion mellan två atomer:
U – interaktionsenergi;
U = U FÖREGÅENDE. + U RETURN
 - Lennard-Jones ekvation
, c, b, m = konst
- Lennard-Jones ekvation
, c, b, m = konst
I fall av interaktion av atomer med en fast yta är det nödvändigt att summera alla interaktioner.
x – avstånd till ytan
r – aktionsradie för attraktionskrafter
dV – volym
n – antal ytmolekyler
U ADS. – energi av adsorptionsinteraktion



Vid adsorption ökar attraktionen. Och i fallet med icke-polär-opolär interaktion är adsorptionen övervägande lokaliserad i fördjupningar.
Elektrostatisk interaktion.
Polär adsorbent – opolärt adsorbat
Icke-polär adsorbent – polärt adsorbat
Polär adsorbent – polärt adsorbat.
M  Adsorbatmolekylen representeras som en dipol, och adsorbenten representeras som en ledare i vilken adsorbatmolekylen inducerar en dipolspegel symmetriskt i förhållande till den givna.
Adsorbatmolekylen representeras som en dipol, och adsorbenten representeras som en ledare i vilken adsorbatmolekylen inducerar en dipolspegel symmetriskt i förhållande till den givna.
X – avstånd till mitten
Vid interaktion uppstår potential:
 ,
,
 - dipolmoment.
- dipolmoment.
Potentialen tenderar att ta på maxvärdet, d.v.s. dipoler tenderar att orientera sig vinkelrätt mot ytan.
Eftersom en ökning av temperaturen främjar tillväxten av Brownsk rörelse, leder det till hämning av adsorptionsprocessen.
Vid elektrostatisk interaktion är adsorbatet övervägande lokaliserat på utsprången.
Grundläggande adsorptionsekvation.
Vid adsorption sker en omfördelning av komponenten, vilket innebär att den kemiska potentialen förändras. Adsorptionsprocessen kan betraktas som övergången av ytenergi till kemisk energi.
Skiktvolym = 0, sedan den generaliserade ekvationen för termodynamikens I- och II-lagar:
T = const; (1) = (2) => 

För ett tvåkomponentsystem:
,
 ,
,
 =>
=>
=>
 - Gibbs adsorptionsekvation
.
- Gibbs adsorptionsekvation
.
För tv-adsorption. kropp - gas: , 
 ,
,

 - isoterm
- isoterm
 - isobar
- isobar
 - isopycnal
- isopycnal
 - isoster
- isoster
Isoterm, isopycne, isostere är släkt med varandra.
Därför att adsorptionsfunktion 



Henry isoterm Langmuir isoterm



Termodynamik. Adsorption.
För kondenserad materia:
 ,
,
 ,
,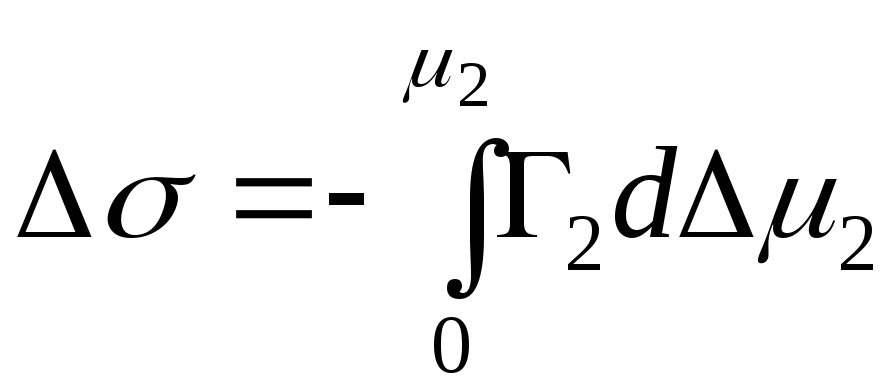
 - integrerad förändring i Gibbs energi
.
- integrerad förändring i Gibbs energi
.

P – tryck över en krökt yta, Р S – tryck över en plan yta
 - adsorptionspotential
- adsorptionspotential
Differentiell förändring i entrapi 
 , Г = konst
, Г = konst
- differentiell entropiförändring
- differentiell entalpi för adsorption
 - isosterisk adsorptionsvärme
- isosterisk adsorptionsvärme
 - kondensationsvärme
- kondensationsvärme
 - nettoadsorptionsvärme
- nettoadsorptionsvärme

 ,
,




Qa – integrerat adsorptionsvärme, 
Qra – integrerad nettoadsorptionsvärme,

Henrys ekvation
Studiet av adsorption kompliceras av ytans heterogenitet, så de enklaste lagarna erhålls för homogena ytor.
Låt oss betrakta växelverkan mellan gaser och en fast yta, när en gas övergår från ett jämviktstillstånd i volymen till ett jämviktstillstånd på ytan. Detta fall är analogt med jämvikten mellan gaser i ett gravitationsfält.
 ,
,
 ,
=>
,
=> -Henrys ekvation
-Henrys ekvation
 - Fördelningskoefficient
- Fördelningskoefficient
Under adsorptionsprocessen sker en förändring i kemiska potentialer.
För bulkfasen: 
För gas på ytan: 
I ett tillstånd av balans  , dvs.
, dvs.

I Henrys ekvation beror konstanten inte på koncentration
Henrys ekvation är giltig i området med låga tryck och koncentrationer. När koncentrationen ökar är två typer av avvikelser från Henrys lag möjliga:
1 – positiva avvikelser, D minskar, A minskar
2 – negativa avvikelser, D – ökar, A – ökar.
Typen av avvikelse bestäms av dominansen av en eller annan typ av adsorbent-adsorbatinteraktion.
Med stark adhesiv interaktion ökar aktivitetskoefficienterna - en positiv avvikelse. Vid kohesiva interaktioner observeras negativa avvikelser.
Monomolekylär adsorption.
Langmuir isoterm.
De enklaste mönstren erhölls i Henrys teori. Langmuir föreslog en teori enligt vilken adsorption betraktas som en kvasikemisk reaktion. Vart i:
Ytan är energimässigt homogen.
Adsorptionen är lokaliserad, varje adsorptionscentrum interagerar med en adsorbatmolekyl.
Adsorbatmolekyler interagerar inte med varandra.
Monolageradsorption.

 - yta,
- yta,  - adsorbat,
- adsorbat,  - adsorptionskomplex.
- adsorptionskomplex.
 , sedan koncentrationen av adsorptionsställen:
, sedan koncentrationen av adsorptionsställen:  ,
, - begränsa adsorptionen.
- begränsa adsorptionen.
 , då är reaktionskonstanten:
, då är reaktionskonstanten: 

 - Langmuirs ekvation.
- Langmuirs ekvation.
Beroende av adsorption på koncentration
1 )
)
 ,
,

2) område med höga koncentrationer
 - begränsande adsorption, bildning av ett monomolekylärt skikt
- begränsande adsorption, bildning av ett monomolekylärt skikt
För Gibbs energi: .
g är entropifaktorn.
I fallet med Henry-isotermen karakteriserar Gibbs-energin övergången av adsorbatet från standardtillståndet i bulken till standardtillståndet på ytan. I fallet med Langmuir-isotermen  kännetecknar graden av affinitet mellan adsorbenten och adsorbatet.
kännetecknar graden av affinitet mellan adsorbenten och adsorbatet.
 hittat från van't Hoff isobar.
hittat från van't Hoff isobar.

 , Då
, Då  , härifrån
, härifrån  .
.

 - grad av ytfyllnad.
- grad av ytfyllnad.




 - antal lediga platser,
- antal lediga platser,  - antal upptagna platser.
- antal upptagna platser.
 ,
,

De där. i området med höga koncentrationer är antalet fria ställen omvänt proportionellt mot mängden adsorbat.
Adsorption av en blandning av gaser på en homogen yta.
I detta fall betraktas adsorptionsprocessen som två parallella reaktioner.
 (1)
(1)

 (2)
(2)


Adsorption av en blandning av gaser på en ojämn yta.
Vid ojämn yta kan man inte begränsa sig till genomsnittliga fyllningar.
Som ett resultat av konkurrens är lokalisering av olika adsorbater möjlig i områden av olika typer.
I detta fall förhållandet  .
.
 ,
,
 - mättat ångtryck hos adsorbatet.
- mättat ångtryck hos adsorbatet.
 ,
,
 - adsorptionsvärme.
- adsorptionsvärme.
"+" - symbatberoende, "-" - antibatberoende, "N" - ingen korrelation.
"+" - adsorption fortskrider enligt samma mekanism. I de mest energimässigt gynnsamma områdena adsorberas gas med hög affinitet till ytan övervägande.
"-" - adsorption sker genom olika mekanismer och fram till en viss tidpunkt finns ingen konkurrens om ytan.
Monomolekylär adsorption realiseras huvudsakligen under den fysiska adsorptionen av gaser vid låga värden sid, såväl som vid vätske/gas-gränsytan.
Polymolekylär adsorption.
BET teori(Brunauer, Emmett, Teller).
I det fall då bildandet av ett monoskikt inte är tillräckligt för att kompensera för ytenergin är adsorption polymolekylär och kan betraktas som ett resultat av påtvingad kondensation under inverkan av ytkrafter.
Nyckelord:
När en adsorbatmolekyl träffar en ockuperad plats bildas en multipel uppsättning.
När vi kommer närmare sid Till sid s antalet fria adsorptionsplatser minskar. Inledningsvis ökar antalet platser som upptas av singel, dubbel etc. för att sedan minska. i set.
På sid =sid s adsorption övergår i kondensation.
Det finns inga horisontella interaktioner.
För det första lagret är Langmuir-isotermen uppfylld.
Ytan betraktas som en uppsättning adsorptionsställen. Villkoret för dynamisk jämvikt är giltigt: kondensationshastigheten på fria platser är lika med avdunstningshastigheten från ockuperade platser.
a är kondensationskoefficienten (fraktionen av molekyler som kondenseras på ytan);
 ,
,
Zm – maximalt antal lediga platser.
 - frekvensen av atomära vibrationer i riktningen vinkelrät mot ytan.
- frekvensen av atomära vibrationer i riktningen vinkelrät mot ytan.
För det första lagret, dynamiska jämviktsförhållanden:



 , Då
, Då
 - Langmuirs ekvation.
- Langmuirs ekvation.
För det andra lagret kommer det att vara sant: 
För det i-te lagret: 
För enkelhetens skull antas det att a och ν är lika för alla lager utom det första. För alla skikt utom det första är adsorptionsvärmen konstant. För det sista lagret är adsorptionsvärmet lika med kondensationsvärmet. Som ett resultat erhölls ekvationen
 (*)
(*)
C- konstant,
När det gäller BET-teori, konstanten MED kännetecknar Gibbs energi av ren adsorption. Ekvationen innehåller bara en konstant, och denna ekvation är också mycket viktig för att bestämma adsorbentens specifika yta.
Eftersom värme frigörs till följd av adsorption bestäms specifika ytareor vid låga temperaturer.
 ????????????
????????????


Den största nackdelen med teorin– försummelse av horisontella interaktioner till förmån för vertikala.
Ekvationen håller i intervallet  från 0,05 till 0,3.
från 0,05 till 0,3.
Var  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 – interaktionen adsorbat – adsorbat påverkas.
> 0,3 – interaktionen adsorbat – adsorbat påverkas.
Redovisning av adsorbat-adsorbat-interaktioner.
Interaktioner uppstår när grenade molekyler eller molekyler adsorberas på en opolär yta. Kan bilda medarbetare. I detta fall ändras formen på adsorptionsisotermerna.
A  adsorbenten är inte polär.
adsorbenten är inte polär.
Diagram 1 motsvarar svaga adsorbat-adsorbat-interaktioner och starka adsorbat-adsorbent-interaktioner.
Diagram 2 motsvarar starka adsorbat-adsorbat och starka adsorbat-adsorbent-interaktioner.
Diagram 3 motsvarar stark adsorbat-adsorbat-interaktion och svag adsorbat-adsorbent-interaktion.
 ,
,

I fallet med interaktion mellan adsorbatmolekyler är det nödvändigt att ta hänsyn till förändringar i aktivitetskoefficienter. Och denna ekvation är skriven som:
 - Frunkin, Fowler, Guggenheims ekvation.
- Frunkin, Fowler, Guggenheims ekvation.
k– attraktionskonstant.
Polyanys potentiella teori.
Denna teori härleder inte någon typ av adsorptionsisoterm, men gör det möjligt att beräkna isotermer vid en annan temperatur.
Adsorption- detta är resultatet av attraktionen av adsorbatet till ytan av adsorbenten på grund av verkan av adsorptionspotentialen, som inte beror på närvaron av andra molekyler och beror på avståndet mellan ytan och adsorbatmolekylen.
 ,
,
 - adsorptionspotential.
- adsorptionspotential.
Eftersom ytan är ojämn ersätts avståndet med adsorptionsvolymen  .Adsorptionsvolymär volymen innesluten mellan ytan och den punkt som motsvarar ett givet värde
.Adsorptionsvolymär volymen innesluten mellan ytan och den punkt som motsvarar ett givet värde  .
.
Adsorptionspotentialär arbetet med att överföra 1 mol adsorbat utanför en given adsorptionsvolym till en given punkt av adsorptionsvolymen (eller arbetet med att överföra 1 mol mättad ånga av ett adsorbat som är i jämvikt med ett flytande adsorbat i frånvaro av en adsorbent till en ångfas i jämvikt med adsorbenten).

Karakteristisk kurva
 - adsorptionspotential,
- adsorptionspotential, 
För en given adsorbent och olika adsorbater gäller följande: 
För olika typer av adsorbater  ,
,
Var  potentialer för adsorptionsisotermer vid relativa tryck
potentialer för adsorptionsisotermer vid relativa tryck  för adsorbat 1 och för adsorbat 2. Detta förhållande är ett konstant värde.
för adsorbat 1 och för adsorbat 2. Detta förhållande är ett konstant värde.
 - affinitetskoefficient
- affinitetskoefficient
Teori om kapillär kondensation.
Förloppet av adsorptionsprocessen beror till stor del på strukturen hos den porösa kroppen.
|
Mikroporös | |
|
Övergångs porös | |
|
Makroporös |
I fallet med mikroporösa sorbenter överlappar fälten av adsorptionskrafter. När det gäller makroporösa sorbenter fungerar porerna som transportkanaler. Kondensationsprocesser är mest betydelsefulla i övergångsvis porösa kroppar. Kapillärkondensering börjar vid vissa värden sid Och  , när en del av ytenergin redan har kompenserats. En nödvändig förutsättning är att ytan ska vara självvätande. Processen beskrivs Thompson-Kelvins ekvation.
, när en del av ytenergin redan har kompenserats. En nödvändig förutsättning är att ytan ska vara självvätande. Processen beskrivs Thompson-Kelvins ekvation.

 - för vätning är krökningscentrum i gasfas.
- för vätning är krökningscentrum i gasfas.
Vid kapillärkondensation har adsorptionsisotermen en hysteretisk form. Den nedre grenen motsvarar adsorptionsprocessen och den övre grenen motsvarar desorptionsprocessen.
Alla typer av porer kan reduceras till tre typer:
|
Konisk |
Cylindrisk med en sluten ände |
Cylindrisk med två öppna ändar |
|
Processfyllning utförs från botten av poren. Adsorptionsisotermen och desorptionsisotermen sammanfaller i detta fall, eftersom adsorptionsprocessen börjar från en sfär och desorptionsprocessen också börjar med att vissa sfärer försvinner.
↓ |
Det finns ingen hysteres. Slag framåt och bakåt beskrivs av ekvationen:
|
Det finns ingen botten någonstans, fyllningen av poren kommer att gå längs cylinderns väggar.
cylinder: Isotermen kommer att ha ett hysteretiskt utseende.
↓ |


 - sfär,
- sfär, ,
,



I  Under väta förhållanden uppstår kondens vid lägre tryck, vilket är energetiskt gynnsamt. Från desorptionsgrenen erhålls porstorleksfördelningskurvor.
Under väta förhållanden uppstår kondens vid lägre tryck, vilket är energetiskt gynnsamt. Från desorptionsgrenen erhålls porstorleksfördelningskurvor.
Differentialkurvans maximum förskjuts åt vänster i förhållande till integralkurvans inflexionspunkt. Den totala volymen av små porer är liten, men har stora ytareor. Med ökande porstorlek ökar deras volym som  , och området är som
, och området är som  På grund av detta observeras en förskjutning i differentialkurvans maximum.
På grund av detta observeras en förskjutning i differentialkurvans maximum.
Adsorption vid gränssnittet mellan fast och vätska.
När det gäller adsorption vid fast-gas-gränssnittet försummade vi en komponent. I fallet med adsorption vid gränsytan mellan fast och vätska, tränger adsorbatet undan lösningsmedelsmolekyler från ytan av adsorbenten.
 ,
,
Ekvationen är korrekt:

 ,
,
N 1, N 2 – molfraktioner av lösningsmedel och komponent, N 1 + N 2 = 1, sedan
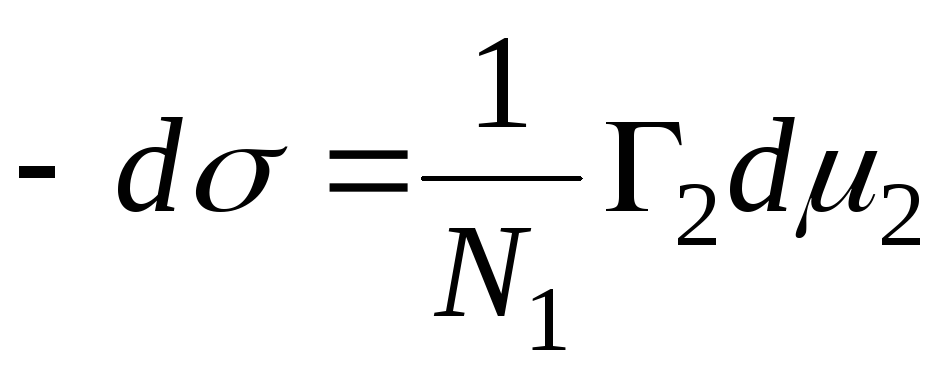 ,
=>
,
=>
 , då är adsorptionsekvationen för gränssnittet fast-vätska.
, då är adsorptionsekvationen för gränssnittet fast-vätska.
Adsorption (G) > 0 at  <
0
<
0
Om värdena  för komponenten och lösningsmedlet är mycket olika, i detta fall beroendet G från N har ett extremum vid värdet N
~ 0,5.
för komponenten och lösningsmedlet är mycket olika, i detta fall beroendet G från N har ett extremum vid värdet N
~ 0,5.
E  om
om  har nära värden, i detta fall kan tecknet på adsorption ändras. Missbruk G från N korsar x-axeln
har nära värden, i detta fall kan tecknet på adsorption ändras. Missbruk G från N korsar x-axeln
Funktion skärningspunkt G(N) med x-axeln kallas adsorptionsazeotrop. Detta innebär att de två komponenterna inte kan separeras på en given adsorbent.
Ekvation för adsorptionsisoterm med utbyteskonstant.
Under adsorption vid gränsytan mellan fast och vätska sker en konstant omfördelning av komponenter mellan ytan av adsorbenten och volymen av lösningen.
 - komponenter (- - hänvisar till ytan)
- komponenter (- - hänvisar till ytan)

 ,
,
 ,
, .
.
 ,
,


Adsorption vid vätske-gas-gränsytan
R  Låt oss betrakta förändringen i koncentrationsprofilen när vätske-gasgränsytan korsas. Låt komponent 2 vara flyktig.
Låt oss betrakta förändringen i koncentrationsprofilen när vätske-gasgränsytan korsas. Låt komponent 2 vara flyktig.
Cs – koncentration i ytskiktet.
Baserat på definitionen av överskottsadsorption

Om komponenten inte är flyktig, kommer adsorptionsvärdet att skrivas enligt följande:
P  ri
ri 

I ekv.  ett ämnes natur beskrivs av dess derivat
ett ämnes natur beskrivs av dess derivat  .
.
Ytspänningsisotermen kan ha formen 1 eller 2:
1 – ytaktiva ämnen
2 – ytaktiva ämnen
Ytaktivitet g är ämnens förmåga att minska ytspänningen i ett system.

 - tjockleken på ytskiktet
- tjockleken på ytskiktet
C s– koncentration av komponenten i ytskiktet
MED– volymkoncentration
För en homolog serie finns en regel:
 - Traubo Duclos regel
- Traubo Duclos regel
För en homolog serie ser adsorptionsisotermen ut så här:
Istället för A skriver vi G, eftersom adsorptionen är överdriven i ytskiktet.
Ytspänningsisoterm:

 - ytspänning av ett rent lösningsmedel.
- ytspänning av ett rent lösningsmedel.
 - fundamental adsorptionsekvation;
- fundamental adsorptionsekvation;
 - Langmuirs ekvation.
- Langmuirs ekvation.
Låt oss lösa dem tillsammans:

- Shishkovskys ekvation.
B– konstant för den homologa serien.
A- när man flyttar från en homolog till en annan ökar med 3-3,5 gånger 
![]()
1 – område med låga koncentrationer
![]()
2 – genomsnittlig koncentration
3 – monomolekylärt skikt
Ytaktiva ämnen är difila molekyler, dvs. inkluderar en polär grupp och en opolär kolväteradikal.
o är den polära delen av molekylen.
| - opolär del av molekylen.
I ett polärt lösningsmedel är ytaktiva molekyler orienterade på ett sådant sätt att den polära delen av molekylen är vänd mot lösningsmedlet och den opolära delen trycks in i gasfasen.
I Shishkovskys ekvation  , är den konstant för den homologiska serien.
, är den konstant för den homologiska serien.
Den ytaktiva effekten börjar synas med n>5. Vid koncentrationer högre än koncentrationen av det monomolekylära skiktet sker micellisering i ytaktiva lösningar.
Micelle– kallas ett aggregat av amfifila ytaktiva molekyler, vars kolväteradikaler bildar en kärna och de polära grupperna omvandlas till vattenfasen.
Micellmassa – micellmassa.
H  antal molekyler – aggregeringsnummer.
antal molekyler – aggregeringsnummer.
Sfäriska miceller
Vid micellisering etableras jämvikt i lösningen
CMC – kritisk koncentration av micellbildning.
Eftersom vi anser att micellen är en separat fas:
För en homologisk serie finns en empirisk ekvation:
a– upplösningsenergi för den funktionella gruppen.
b – ökning av adsorptionspotential, adsorptionsarbete per metylenenhet.
– ökning av adsorptionspotential, adsorptionsarbete per metylenenhet.
Närvaron av en kolvätekärna i miceller skapar möjligheten för föreningar som är olösliga i vatten att lösas i vattenlösningar av ytaktiva ämnen; detta fenomen kallas solubilisering (det som löser sig är solubiliseringsmedlet, det ytaktiva ämnet är solubiliseringsmedlet).
Slammet kan vara helt opolärt, kan innehålla både polära och opolära delar och kommer att vara orienterat som en ytaktiv molekyl.
I vilket fall som helst, under solubilisering sker en ökning av micellmassa och aggregationsantal, inte bara på grund av inkluderingen av solubilisat, utan också på grund av en ökning av antalet ytaktiva molekyler som är nödvändiga för att upprätthålla ett jämviktstillstånd.
Solubilisering är effektivare ju lägre molekylvikten solubilisatet har.

 ~ 72 mN\m.
~ 72 mN\m.
 ~ 33 mN\m.
~ 33 mN\m.
Effektiviteten hos ytaktiva ämnen beror på CMC-värdet.
2D Ytskiktstryck

→ -ytspänningskrafter.
- tvådimensionellt tryck.
Ytskiktet är en kraft lika med skillnaden i ytspänning för en lösning av ytaktivt medel och ett rent lösningsmedel, riktad mot en ren yta.
En jämvikt upprättas mellan lösningen och ytskiktet



På  det finns ett område där
det finns ett område där  beror linjärt på koncentration.
beror linjärt på koncentration.
G [mol/m2].
 -yta som upptas av en mol av ett ämne
-yta som upptas av en mol av ett ämne
Då kommer den tvådimensionella tryckisotermen att ha formen 
 - tvådimensionell tryckisoterm.
- tvådimensionell tryckisoterm.
Missbruk  från S M:
från S M:

På  - det tvådimensionella trycket ökar kraftigt. På
- det tvådimensionella trycket ökar kraftigt. På  tvådimensionell är deformerad, vilket orsakar plötslig tillväxt
tvådimensionell är deformerad, vilket orsakar plötslig tillväxt  .
.
En film avgränsad av identiska faser på båda sidor kallas dubbelsidig. I sådana filmer observeras konstant rörelse av moderluten.
Filmer som är mindre än 5 nm tjocka kallas svarta filmer.

Adsorptionsskikt måste ha två egenskaper: viskositet och lätt rörlighet, flytbarhet och elasticitet.
Marangoni-effekten är självläkande.
Gibbs triangel,  - övertryck.
- övertryck.
Filmen har töjts och på grund av att en del av vätskan har lämnat rusar de ytaktiva ämnena in i det fria utrymmet. Gibbs triangel.
Effekten av kroppars adsorptionsstyrka.
Det finns alltid ett adsorptionsskikt på ytan av filmen, för vilket då
Langmuirs ekvation:


 till tvådimensionellt tryck
till tvådimensionellt tryck
 - en analog till Shishkovsky-ekvationen
- en analog till Shishkovsky-ekvationen
Elektrokinetiska fenomen. Elektriskt dubbelskikt (EDL).
Gelemholtz modell. Gouy-Chapman teori.
1808 flygning

U – formade rör, doppa 2 elektroder i det. Lagen om kommunicerande kärl överträds och en förändring av vätskenivån i röret inträffar - elektrokinetiska fenomen.
Kinetiska fenomen:
Elektrofores
Elektroosmos
Flödes (flöde) potential
Sedimentationspotential
1 och 2 uppstår när en potentialskillnad appliceras, 3 och 4 orsakar stansning och sedimentering av kolloidala partiklar uppkomsten av en potentialskillnad.
Elektroosmos är rörelsen av ett dispersionsmedium i förhållande till en stationär dispergerad fas under inverkan av en elektrisk ström.
Elektrofores – detta är rörelsen av dispergerade faspartiklar i förhållande till ett stationärt dispersionsmedium under påverkan av en elektrisk ström.
P  Anledningen till förekomsten av elektrokinetiska fenomen är den rumsliga separationen av laddningar och uppkomsten av ett dubbelt elektriskt lager.
Anledningen till förekomsten av elektrokinetiska fenomen är den rumsliga separationen av laddningar och uppkomsten av ett dubbelt elektriskt lager.
Det elektriska dubbelskiktet är en platt kondensator, en platta är bildad av potentialbestämmande joner, den andra av motjoner. Jonerna kontamineras på samma sätt som potentialbestämmande kojoner trycks in i lösningens volym. Avstånd mellan plattorna  . Potentialen sjunker linjärt, potentialskillnaden
. Potentialen sjunker linjärt, potentialskillnaden  .
.
En extern potentialskillnad orsakar uppkomsten av en skjuvmodul  är ett par krafter per ytenhet som verkar längs ytan av en fast kropp.
är ett par krafter per ytenhet som verkar längs ytan av en fast kropp.

I jämvikt är skjuvmodulen lika med den viskösa friktionsmodulen (  ).
).

I våra förhållanden  ,
,

 - Gelemholtz-Smalukowskis ekvation
- Gelemholtz-Smalukowskis ekvation
 - linjär hastighet för fasförskjutning.
- linjär hastighet för fasförskjutning.
E– elektrisk fältstyrka.
 - potentialskillnad mellan plattor
- potentialskillnad mellan plattor
 - elektroforetisk rörlighet [m2/(V*s)].
- elektroforetisk rörlighet [m2/(V*s)].
Helemholtz-modellen tar inte hänsyn till molekylers termiska rörelse. I verkligheten är fördelningen av joner i dubbelskiktet mer komplex.
Gui och Chapman identifierade följande orsaker till DES:
Övergången av en jon från en fas till en annan när jämvikt är etablerad.
Jonisering av fast fasmaterial.
Komplettering av ytan med joner närvarande i dispersionsmediet.
Polarisering från en extern strömkälla.
Det elektriska dubbelskiktet har en luddig eller diffus struktur. Jonerna tenderar att vara jämnt fördelade i det diffusa skiktet.
Det diffusa skiktet består av motinoner, längden på skiktet bestäms av deras kinetiska energi. Vid temperaturer som närmar sig absolut noll är motjoner så nära potentialbestämmande joner som möjligt.
Danyas teori bygger på två ekvationer:
Boltzmanns ekvation

 - arbeta mot krafterna från elektrostatisk interaktion.
- arbeta mot krafterna från elektrostatisk interaktion.
 - volymetrisk laddningstäthet.
- volymetrisk laddningstäthet.

Poissons ekvation 
Eftersom tjockleken på EDL är mycket mindre än partikelstorleken och för en platt EDL derivatan med avseende på koordinater  Och
Och  är avskaffad.
är avskaffad. 
För e y at y<<1 функцию можно разложить в ряд Маклорена:

Låt oss då begränsa oss till två termer i serien:


 - DEL-tjocklek är det avstånd från vilket DEL-potentialen minskar e en gång.
- DEL-tjocklek är det avstånd från vilket DEL-potentialen minskar e en gång.
Ju lägre temperatur, desto mindre  . Vid T→0 – platt DEL. Ju högre koncentration, desto mer jag, desto mindre
. Vid T→0 – platt DEL. Ju högre koncentration, desto mer jag, desto mindre  .
.





 "–" betyder att potentialen minskar med avståndet. =>
"–" betyder att potentialen minskar med avståndet. =>
=>

 ,
,
 - potentialen minskar exponentiellt.
- potentialen minskar exponentiellt.
Potential för ytladdningstäthet: 
Ytladdning är en volymladdning med motsatt tecken, integrerad över avstånd.




=>


Där potentialen minskar med 2,7 gånger - 
Dubbla lagerkapacitet 
Nackdelen med teorin är att närvaron av Helemholtz-skiktet inte beaktas, d.v.s. tar inte hänsyn till  , därav felen vid bestämning av huvudparametrarna. Det förklarar inte heller inverkan av joner av olika natur på tjockleken av det elektriska dubbelskiktet.
, därav felen vid bestämning av huvudparametrarna. Det förklarar inte heller inverkan av joner av olika natur på tjockleken av det elektriska dubbelskiktet.
Sterns teori. Struktur av en kolloidal micell.
Det elektriska dubbelskiktet består av två delar: tätt och diffust. Ett tätt skikt bildas som ett resultat av interaktionen av potentialbildande joner med specifikt adsorberade. Dessa joner är som regel delvis eller helt uttorkade och kan ha antingen samma eller motsatt laddning till de potentialbestämmande jonerna. Det beror på förhållandet mellan elektrostatisk interaktionsenergi  och specifik adsorptionspotential
och specifik adsorptionspotential  . De täta skiktjonerna är fixerade. Den andra delen av jonerna ligger i det diffusa skiktet, dessa joner är fria och kan röra sig djupare in i lösningen, d.v.s. från ett område med högre koncentration till ett område med lägre koncentration. Den totala laddningstätheten består av två delar.
. De täta skiktjonerna är fixerade. Den andra delen av jonerna ligger i det diffusa skiktet, dessa joner är fria och kan röra sig djupare in i lösningen, d.v.s. från ett område med högre koncentration till ett område med lägre koncentration. Den totala laddningstätheten består av två delar.

 -laddning av Helmholtzskiktet
-laddning av Helmholtzskiktet
 -Diffus lagerladdning
-Diffus lagerladdning
Ytan har ett visst antal adsorptionscentra, som var och en interagerar med en motjon. Konstanten för en sådan kvasikemisk reaktion är lika med:

 , Var
, Var  - molfraktion av motjoner i lösning
- molfraktion av motjoner i lösning
Helmholtz distribution
Potentialen minskar linjärt
Gouy potentiell distribution. Det finns inget tätt lager, potentialen minskar exponentiellt från värdet 
Sternfördelning.
Inledningsvis är potentialminskningen linjär och sedan exponentiell.
När ett elektriskt fält appliceras i fallet med elektrofores är det inte partikeln i den fasta fasen som rör sig direkt, utan partikeln i den fasta fasen med ett lager av joner som omger den. DES upprepar formen av den dispergerade faspartikeln. När en potential appliceras rivs en del av det diffusa lagret av. Brytlinjen kallas glidande gräns.
Potentialen som uppstår vid glidgränsen som ett resultat av separation av en del av det diffusa skiktet kallas elektrokinetisk potential(Zeta potential  ).
).
En dispergerad faspartikel med ett omgivande skikt av motjoner och ett dubbelt elektriskt skikt kallas micell.
Regler för att skriva kolloidala miceller:


1-1 laddningselektrolyt
T – dispergerad faspartikel.
AA är gränsen mellan de täta och diffusa delarna.
BB – glidande gräns.
Den glidande gränsen kan eller kanske inte sammanfaller med linje AA.
pH-värdet vid vilket zetapotentialen är noll kallas isoelektrisk punkt.
CaCl2 + Na2SO4 → CaSO4 ↓ + 2NaCl
1. Överskott av CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n - x)Cl-) 2 x + x Cl - - micell notation.
CaSO 4 m – ballast.
CaSO 4 m∙nCa 2+ – kärna.
CaSO 4 m∙nCa 2+ 2( n - x)Cl - - partikel.
2. Överskott av Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO4 m∙nSO4 2- 2(n-x)Na+) 2x- 2xNa+ - micell
CaSO 4 m – ballast.
CaSO 4 m∙nSO 4 2 + – kärna.
CaS04 m∙nSO4 2-2(n-x)Na+-partikel
Gelemholtz-Smoluchowskis ekvation

 - linjär hastighet för gränsförskjutning (vid elektroosmos).
- linjär hastighet för gränsförskjutning (vid elektroosmos).
 - potentialskillnad över kondensatorplattorna (vid elektroosmos).
- potentialskillnad över kondensatorplattorna (vid elektroosmos).


 - lösningens volymetriska flödeshastighet, S– cellens tvärsnittsarea.
- lösningens volymetriska flödeshastighet, S– cellens tvärsnittsarea.

E– elektrisk fältstyrka.


 (för elektroosmos).
(för elektroosmos).
För flödespotential:

 - potential
- potential
 - tryck på membranet
- tryck på membranet
Som regel är värdena för elektroforetiska rörligheter och elektroosmotiska rörligheter mindre än de beräknade. Detta händer på grund av:
Avslappningseffekt (när en dispergerad faspartikel rör sig bryts symmetrin i jonatmosfären).
Elektroforetisk hämning (förekomsten av ytterligare friktion som ett resultat av rörelsen av motjoner).
Förvrängning av strömledningar vid elektriskt ledande partiklar.
Samband mellan ytspänning och potential. Lippmanns ekvation.
Bildandet av EDL sker spontant på grund av systemets önskan att minska sin ytenergi. Under konstanta förhållanden T Och sid den generaliserade ekvationen för termodynamikens första och andra lag ser ut så här:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1:a Lippmann-ekvationen.
- 1:a Lippmann-ekvationen.
 - ytladdningstäthet.
- ytladdningstäthet.
 - differentiell kapacitans.
- differentiell kapacitans.
 - 2:a Lippmann-ekvationen.
- 2:a Lippmann-ekvationen.
MED– kapacitet.
Låt oss lösa den första Lippmann-ekvationen och den fundamentala adsorptionsekvationen:
 ,
,


 , Då
, Då
 - Nernsts ekvation
- Nernsts ekvation
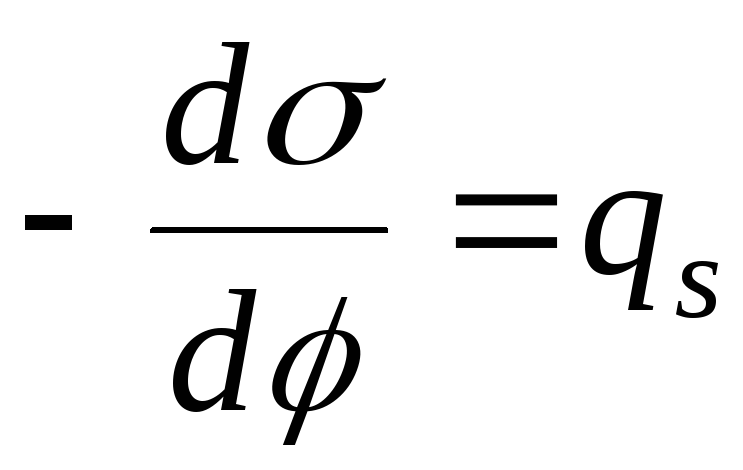 ,
,
 ,
,




 - ekvation för den elektrokapillära kurvan (ECC).
- ekvation för den elektrokapillära kurvan (ECC).
I  :
: , Men
, Men 
Katjoniska ytaktiva ämnen (CPAS) reducerar den katodiska grenen av EKC.
Anjoniska ytaktiva ämnen (APS) reducerar den anodiska grenen av EKC.
Nonjoniska ytaktiva ämnen (NSAS) minskar den mellersta delen av ECC.
Stabilitet av spridda system. Skiftande tryck.
Dispergerade system kan delas in:
System som är termodynamiskt instabila kan vara kinetiskt stabila på grund av övergången till ett metastabilt tillstånd.
Det finns två typer av stabilitet:
Sedimentationsstabilitet (relativt tyngdkraften).
Aggregativ stabilitet. (relativt vidhäftning)
Koaguleringär en process av partikelvidhäftning, vilket leder till förlust av aggregationsstabilitet. Koagulering kan orsakas av temperaturförändringar, pH, omrörning och ultraljud.
Koagulering särskiljs:
Reversibel.
Irreversibel.
Koagulering sker med införandet av elektrolyter.
Koaguleringsregler:

Filma- detta är den del av systemet som ligger mellan två gränsytor.
Skiftande tryck uppstår när filmtjockleken minskar kraftigt som ett resultat av samverkan mellan närmande ytskikt.

"-" - när filmtjockleken minskar, ökar det lösgörande trycket.
P 0 är trycket i bulkfasen, som är en fortsättning på mellanskiktet.
P 1 – tryck i filmen.
Teori om stabilitet. DLFO (Deryagin, Landau, Fairway, Overbeck).
Enligt DLFO-teorin har sönderdelningstryck två komponenter:
Elektrostatisk PE (positiv, det beror på krafterna från elektrostatisk repulsion). Motsvarar en minskning av Gibbs-energin med ökande filmtjocklek.
Molekyl P M (negativ, på grund av verkan av attraktionskrafter). Det orsakas av filmkompression på grund av kemiska ytkrafter, krafternas verkningsradie är tiondelar av nm med en energi på cirka 400 kJ/mol.
Total interaktionsenergi:

 - systemet är aggregerat stabilt
- systemet är aggregerat stabilt
 - instabilt system
- instabilt system
P  positiv komponent.
positiv komponent.
Ökningen beror på en ökning av potentiell energi när tunna filmer komprimeras. För filmer med stor tjocklek kompenseras överskottet av jonenergin och är lika med energiinteraktionen i dispersionsmediets volym.
Om  (
( - film tjocklek,
- film tjocklek,  - jonradie) förtunning av filmen leder till att molekyler och joner försvinner och reduceras med minimal ytenergi i den. Antalet angränsande partiklar minskar, vilket resulterar i att den potentiella energin hos de partiklar som finns kvar i filmen ökar.
- jonradie) förtunning av filmen leder till att molekyler och joner försvinner och reduceras med minimal ytenergi i den. Antalet angränsande partiklar minskar, vilket resulterar i att den potentiella energin hos de partiklar som finns kvar i filmen ökar.


DLVO-teorin betraktar interaktionen mellan partiklar som interaktionen mellan plattor.

Partiklar interagerar inte



 - Laplace ekvation,
- Laplace ekvation,  ,
,


För svagt laddade ytor
För mycket laddade ytor:

Den molekylära komponenten är interaktionen mellan två atomer:
 ~
~
Interaktion mellan en atom och en yta:

Låt oss ta två rekord:
D  För att erhålla den molekylära komponenten är det nödvändigt att summera alla interaktionsenergier för atomerna på den högra och vänstra plattan.
För att erhålla den molekylära komponenten är det nödvändigt att summera alla interaktionsenergier för atomerna på den högra och vänstra plattan.
Var  - Hamaker konstant (tar hänsyn till naturen hos interagerande kroppar).
- Hamaker konstant (tar hänsyn till naturen hos interagerande kroppar).
Den där. interaktionsenergin för partiklar i ett system kan uttryckas med hjälp av potentialkurvor.
I – primär potential minimum. Detta är en zon av irreversibel koagulering, attraktionskrafterna råder.
II – zon av aggregativ stabilitet, frånstötande krafter dominerar.
III – sekundär potentialminimum (eller flockningszon). Det finns ett elektrolytskikt mellan partiklarna i den dispergerade fasen, och partiklarna kan separeras och överföras till zonen för aggregationsstabilitet.

Kurva 1 – systemet är aggregativt stabilt.
Kurva 2 – stabil i zon I, instabil i zon II.
Kurva 3 – koagulering har inträffat i systemet.
Kurva 4 – vid punkt 4 den totala interaktionsenergin U=0,  , motsvarar denna extrema punkt början av snabb koagulering.
, motsvarar denna extrema punkt början av snabb koagulering.
Det finns två fall:
1. Lätt laddade ytor:
U = U E + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - detta är tjockleken på skiktet som motsvarar början av koaguleringsprocessen.
- detta är tjockleken på skiktet som motsvarar början av koaguleringsprocessen.






 - för svagt laddade ytor
- för svagt laddade ytor
 Sedan
Sedan 
2. För mycket laddade ytor:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Låt oss kvadrera (3)

Koagulering:
Vid specifik adsorption kan joner adsorberas i superekvivalenta mängder så att ytan kan ändra sin laddning. Ytan laddas upp igen.
I fallet med specifik adsorption kan joner inte bara av motsatta tecken adsorberas utan även av samma tecken.
Om joner med samma tecken som ytan adsorberas, kommer det i ytskiktet inte att finnas en droppe i potentialen, utan en ökning av den.

Neutraliseringskoagulering (förekommer med deltagande av svagt laddade partiklar och beror inte bara på laddningen av elektrolytkoagulatorn utan också på potentialen vid gränsen för de täta och diffusa skikten).
Smoluchowskis teori om snabb koagulation.

Beroende av koagulationshastighet på elektrolytkoncentration.
I – koagulationshastigheten är låg,
II – koagulationshastigheten är nästan proportionell mot elektrolytkoncentrationen.
III – område med snabb koagulering, hastigheten är praktiskt taget oberoende av koncentrationen.
Grundläggande bestämmelser:
Den initiala solen är monodispers, liknande partiklar har en sfärisk form.
Alla partikelkollisioner är effektiva.
När två primära partiklar kolliderar bildas en sekundär partikel. Sekundär + primär = tertiär. Primär, sekundär, tertiär – mångfald.
När det gäller kemisk kinetik kan koaguleringsprocessen beskrivas med ekvationen:

Lösningen blir ekvationen: 
 - halv koagulationstid. Detta är den tid under vilken antalet solpartiklar minskar med 2 gånger.
- halv koagulationstid. Detta är den tid under vilken antalet solpartiklar minskar med 2 gånger.

 ,
,
 ,
,
 ,
,


När multipliciteten ökar skiftar koagulationskurvornas maximum mot större värden  .
.
Brister:
Antagande om monodispersitet.
Antagande om effektiviteten av alla kollisioner.