Termodynamikk av adsorpsjonsprosesser.
| Parameternavn | Betydning |
| Artikkel emne: | Termodynamikk av adsorpsjonsprosesser. |
| Rubrikk (tematisk kategori) | utdanning |
Grunnleggende definisjoner og metoder for klassifisering av adsorpsjonsprosesser.
Adsorpsjon refererer til fenomener som oppstår på grunn av en spontan reduksjon i overflateenergi.
Adsorpsjon– prosessen med spontan reversibel eller irreversibel omfordeling av komponentene i et heterogent system mellom overflatelaget og volumet av den homogene fasen.
I flerkomponentsystemer overføres fortrinnsvis den komponenten som kraftigere reduserer grenseflatespenningen til overflatelaget. I enkomponentsystemer, under dannelsen av overflatelaget, oppstår en endring i strukturen (en viss orientering av atomer og molekyler, polarisering), kalt autoadsorpsjon.
Den tettere fasen som adsorpsjonsinteraksjoner er lokalisert på kalles adsorbent. Stoffet omfordelt mellom volumet av den homogene fasen og overflatelaget er betegnet med begrepet '' adsorbatʼʼ.
I noen tilfeller er adsorpsjonsprosessen reversibel. I dette tilfellet, under visse forhold, kan en del av de adsorberte molekylene som følge av molekylære kinetiske fenomener bevege seg fra overflatelaget til bulkfasen. Den omvendte prosessen med adsorpsjon kalles desorpsjon.
Metoder for klassifisering av adsorpsjonsprosesser.
Klassifisering av adsorpsjonsprosesser i henhold til tilstanden til aggregering av interagerende faser. Tatt i betraktning avhengigheten av den samlede tilstanden til tilstøtende faser, skilles følgende typer adsorpsjonsprosesser ut:
Adsorpsjon av gasser på faste adsorbenter;
Adsorpsjon av oppløste stoffer ved grensesnittene "fast-væske" og "væske-væske";
Adsorpsjon av overflateaktive stoffer ved væske-gass-grensesnittet.
Klassifisering av adsorpsjonsprosesser i henhold til mekanismen for interaksjon mellom adsorbenten og adsorbatet. Adsorpsjon kan betraktes som interaksjonen av adsorbatmolekyler med de aktive sentrene til adsorbenten. I henhold til mekanismen for deres interaksjon er følgende typer adsorpsjon delt inn:
1) fysisk (molekylær) adsorpsjon– samspillet mellom molekylene til adsorbatet og adsorbenten utføres på grunn av van der Waals-krefter, hydrogenbindinger (uten kjemiske reaksjoner);
2) kjemisk adsorpsjon (kjemisorpsjon)- festingen av adsorbatmolekyler til de aktive sentrene til adsorbenten skjer som et resultat av kjemiske reaksjoner av forskjellige typer (med unntak av ionebytterreaksjoner);
3) ionebytteradsorpsjon (ionebytter) – omfordeling av adsorbatstoffet mellom løsningen og den faste fasen (ionebytteren) i henhold til mekanismen for ionebytterreaksjoner.
For å kvantitativt beskrive adsorpsjonsprosesser brukes to mengder.
1) Absolutt adsorpsjon– mengde (mol) eller masse (kg) av adsorbat per enhet overflateareal eller masse av adsorbenten. Betegnelse – A; dimensjon: mol/m2, mol/kg, kg/m2, kg/kᴦ.
2) Gibbs (overflødig) adsorpsjon– overskudd av adsorbatstoff i et overflatelag av en viss tykkelse sammenlignet med dets mengde i volumet av den homogene fasen, per enhet overflateareal eller masse av adsorbenten. Betegnelse – G; dimensjon: mol/m 2, mol/kᴦ.
Forholdet mellom absolutt og overflødig adsorpsjon kan illustreres ved å bruke ligningen:
Г = А – с * h (3,1)
hvor c er likevektskonsentrasjonen av stoffet i volumet av fasen, mol/m3;
h er tykkelsen på overflatelaget, konvensjonelt antatt å være 10 -9 m.
I multikomponent heterogene systemer, når en eller annen komponent er omfordelt mellom volumet av den homogene fasen og overflatelaget, er ligningen for overflødig indre energi til overflaten gyldig:
U = T * S + s * s + Sm i * n i (3,2)
Ved å redusere alle ledd i ligningen til enhetsareal av interfaseoverflaten, får vi:
U s = T * S s + s + Sm i * Г i (3.3)
hvor Г i = n i / s er overskuddet av den i-te komponenten i overflatelaget, det vil si Gibbs adsorpsjon.
For et enkomponentsystem vil ligning (3.3) ha formen:
G s = s + m * G (3,4)
hvor G s = U s - T * S s – Gibbs energi av overflaten eller arbeidet med å skape en enhetsoverflateareal;
m * G – komprimering av stoffet til det adsorberte stoffet i overflatelaget.
Basert på ligning (3.4) kan vi konkludere med at under adsorpsjon består arbeidet med å lage en interfaseoverflate av arbeidet med overflatedannelse (bryte kohesive bindinger i volumet av adsorbatfasen) og komprimering av stoffet i overflatelaget.
I en tilstand av dynamisk likevekt mellom adsorbenten og adsorbatet, endringen i Gibbs-energien til det heterogene systemet ΔG = 0, er termodynamikken til adsorpsjonsprosessen beskrevet ved ligningen kalt Gibbs fundamentale adsorpsjonsligning:
Ds = SГ i * dm i (3,5)
Denne ligningen er universell, da den er gyldig for alle typer adsorpsjonsprosesser
Spesielle tilfeller av Gibbs adsorpsjonsligning.
1) Adsorpsjon fra løsninger.
For det kjemiske potensialet til den i-te komponenten av systemet under adsorpsjon ved grensesnittene "væske – fast adsorbent" og "væske - gass", er følgende ligninger gyldige:
m i = m i 0 + R*T*ln a i (3,6)
dm i = R*T* d ln a i (3,7)
hvor m i 0 er det kjemiske potensialet til den i-te komponenten av systemet under standardbetingelser;
a i er aktiviteten til den i-te komponenten av systemet under standardforhold.
Basert på dette tar Gibbs adsorpsjonsligning formen:
Г i = - a i / R*T * (ds / da i) (3.8)
For løsninger av ikke-elektrolytter tar vi a i = c i, da:
Г i = - с / R*T * (ds / dс) (3.9)
For elektrolyttløsninger:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
hvor с ± er den gjennomsnittlige ioniske konsentrasjonen av løsningen;
n er den støkiometriske koeffisienten.
2) Adsorpsjon av stoffer fra gassfasen.
I samsvar med Mendeleev-Clayperon-ligningen:
Р = с * R*T (3,11)
I denne forbindelse er Gibbs-ligningen for adsorpsjon av gasser på faste adsorbenter skrevet i følgende form:
Г i = - Р / R*T * (ds / dР) (3.12)
I praksis tillater Gibbs adsorpsjonsligning, basert på overflatespenningsmålinger ved forskjellige verdier av væskekonsentrasjon eller likevektsgasstrykk, å beregne mengden adsorpsjon av stoffer i grenseflatelaget som overflatespenningen er bestemt for.
Termodynamikk av adsorpsjonsprosesser. - konsept og typer. Klassifisering og funksjoner i kategorien "Termodynamikk av adsorpsjonsprosesser." 2017, 2018.
Nåværende side: 6 (boken har totalt 19 sider) [tilgjengelig lesepassasje: 13 sider]
Font:
100% +
34. Arten av adsorpsjonskrefter
Samspillet mellom adsorbentmolekyler med overflaten av adsorbenten under den såkalte. fysisk adsorpsjon kan skyldes ulike årsaker. Da kan potensialet som bestemmer interaksjonen mellom ett adsorbentmolekyl og ett atom i en ikke-polar adsorbent uttrykkes som følger:
θ = −Cr 6 +Br 12 ,
hvor r er avstanden mellom sentrene til partikler; C – dispersjonsattraksjonskonstant; B er en konstant som karakteriserer energien til frastøtende krefter.
Det er ganske åpenbart at på relativt fjerne avstander bør tiltrekningskreftene råde, og på nære avstander bør frastøtningskreftene råde. Også på visse avstander må disse kreftene være like, noe som vil tilsvare et minimum av fri energi. Men det er viktig å merke seg at under adsorpsjon virker dispersjonskrefter samtidig mellom hver ikke-polar partikkel.
Siden interaksjonsenergien til partikler raskt kan avta med avstanden, er det tilstrekkelig for å bestemme potensialet til adsorpsjonskrefter å utføre summering på de nærmeste atomene i adsorbenten. Det er viktig at under adsorpsjonen av komplekse ikke-polare molekyler kan den potensielle energien beregnes omtrentlig som summen av alle potensielle adsorpsjonsenergier til molekylenhetene.
Hvis adsorbenten består av ioner, kan virkningen av de allerede kjente dispersjonskreftene suppleres med virkningen av de induktive tiltrekningskreftene til dipoler som induseres i adsorbentens molekyler av det elektriske feltet, som igjen er skapt av ionene i det adsorberende gitteret.
Med en slik interaksjon kan andelen av induktive krefter i adsorpsjonsinteraksjonen være proporsjonal med polariserbarheten til adsorbentmolekylet og kvadratet av feltstyrken på denne adsorbentoverflaten.
Hvis adsorpsjonen av polare molekyler av adsorbenten skjer på en polar adsorbent, polariserer dipolene i dette tilfellet atomene til adsorbenten, det vil si at de ser ut til å indusere elektriske momenter i dem. På grunn av denne påvirkningen legges den induktive interaksjonen til dispersjonsinteraksjonen.
Den induktive interaksjonen i seg selv er vanligvis liten og kan, avhengig av dipolen til adsorbentmolekylet og polariserbarheten til adsorbenten, nå store verdier. Dersom molekyler adsorberes på en adsorbent som har ioner eller dipoler på overflaten, vil den s.k. interaksjon av ioner eller dipoler av adsorbenten med det elektrostatiske feltet til selve adsorbenten.
I dette tilfellet kan molekylene til adsorbenten til og med være orientert i feltet til adsorbenten, og orienterende Coulomb-interaksjon oppstår. Det skjer vanligvis at energiene til induktive og orienterende interaksjoner er mindre enn energien til dispersive interaksjoner, og derfor er det akseptert at energien til intermolekylær tiltrekning bestemmes av energien til dispersiv tiltrekning.
Adsorpsjon kan også være forårsaket av dannelsen av en hydrogenbinding. En binding av denne typen kan oppstå under adsorpsjon på adsorbenter som inneholder hydroksylgrupper av molekyler som vann, alkoholer, ammoniakk og aminer på overflaten. Når en hydrogenbinding dannes, kan interaksjonsenergien mellom adsorbenten og adsorbenten være ganske stor, og varmen som frigjøres under slik adsorpsjon er betydelig større enn adsorpsjonsvarmen til stoffer som er like i form og størrelse på molekyler. , men danner ikke en hydrogenbinding.
Det er viktig å merke seg at når man kjenner til den termodynamiske beskrivelsen av overflatelaget ved adsorbent-adsorbent-grensesnittet, dets struktur, arten av ulike typer krefter og dynamikken i prosessen, kan man gå videre til studiet av mer kompleks adsorpsjon prosesser.
35. Adsorpsjon som spontan konsentrasjon ved grenseflaten av stoffer som reduserer grenseflatespenningen
Overflateaktive stoffer er delt inn i to store grupper: aktive og inaktive stoffer.
Overflateaktive stoffer er i stand til å samle seg i overflatelaget, og positiv adsorpsjon oppstår G > 0.
Disse typer stoffer må ha en overflatespenning, som igjen må være mindre enn overflatespenningen til løsemidlet, ellers vil opphopningen av stoffet i overflatelaget være ugunstig, og må ha relativt lav løselighet. Med tilstrekkelig god løselighet har overflateaktive molekyler en tendens til å bevege seg fra overflaten og inn i dypet av løsningen. Følgelig vil overflateaktive midler fortrinnsvis presses ut av hoveddelen av væsken til overflaten.
Men med akkumulering av stoffer ved grensen til løsningen i molekylene til disse stoffene, som samhandler svakt med hverandre, vil den intermolekylære interaksjonen i overflatelaget avta, og overflatespenningen vil falle.
Overflateaktive stoffer i forhold til det vandige laget er mange typer organiske forbindelser, fettsyrer med et ganske stort hydrokarbonradikal, salter av disse syrene (såper), sulfonsyrer og deres salter, samt ulike typer alkoholer og aminer. Et karakteristisk trekk ved de fleste molekyler er deres difilisitet: molekylet består av to deler av en polar gruppe og et ikke-polart hydrokarbonradikal. En polar gruppe som har et betydelig dipolmoment og er svært hydrerende kan bestemme det overflateaktive stoffets affinitet for det vandige miljøet. Men hydrokarbonradikalet er årsaken som reduserer løseligheten til disse forbindelsene.
Overflate-inaktive overflateaktive stoffer- disse typer stoffer har en tendens til å forlate overflaten av væsken i sitt volum, noe som resulterer i den såkalte. negativ adsorpsjon G < 0. Поверностно-инактивные вещества также обладают значительным поверхностным натяжением, значительно большим, чем натяжение у растворителя (иначе эти вещества способны самопроизвольно накапливаться в поверхностном слое), также обладают высокой растворимостью, что способствует их стремлению уйти с поверхности жидкости в объем. Взаимодействие между молекулами поверхностно-инактивного вещества и растворителя всегда больше, чем взаимодействие между самими молекулами растворителя, поэтому они и стремятся перейти в объем раствора. Overflateinaktive stoffer I forhold til vann er det mange uorganiske elektrolytter: syrer, alkalier, salter. Overflateaktive molekyler har ikke en hydrofob del og kan desintegreres i vann til svært hydratiserende ioner.
Eksempler Overflateaktive stoffer er også noen organiske forbindelser der den ikke-polare delen av molekylet er fraværende eller svært liten. Disse stoffene inkluderer maursyre og aminoeddiksyrer.
I ikke-vandige løsningsmidler kan uorganiske elektrolytter også øke overflatespenningen, avhengig av løsningsmidlet.
For eksempel Når natriumjodid innføres i metanol, øker overflatespenningen kraftig; for etanol er overflatespenningen omtrent 2 ganger større. Overflateaktiviteten til stoffer kan avhenge ikke bare av stoffets natur, men også av løsningsmidlets egenskaper. Hvis et løsemiddel har høy overflatespenning, kan det oppløste stoffet vise betydelig overflateaktivitet.
36. Adsorpsjonsteorier
La oss vurdere de vanligste adsorpsjonsteoriene som beskriver visse typer adsorpsjon ved grensesnittet "fast gass" eller "fast løsning".
Teorien om monomolekylær adsorpsjon av I. Langmuir.
1. Adsorpsjon er lokalisert og forårsakes av krefter nær kjemiske.
2. Adsorpsjon skjer bare på aktive sentre - fremspring eller fordypninger på overflaten av adsorbenten, preget av tilstedeværelsen av frie valenser. De aktive sentrene anses som uavhengige og identiske.
3. Hvert aktivt senter er i stand til å samhandle med bare ett adsorbatmolekyl; Bare ett lag med adsorberte molekyler kan dannes på overflaten.
4. Adsorpsjonsprosessen er reversibel og i likevekt; det adsorberte molekylet beholdes av det aktive stedet i noen tid, hvoretter det desorberes; Etter en tid etableres dynamisk likevekt.
Maksimal mulig adsorpsjonsverdi G o oppnås forutsatt at alle aktive sentre er okkupert av adsorbatmolekyler. Monomolekylær adsorpsjon isoterm ligning som relaterer størrelsen på adsorpsjonen G med adsorbatkonsentrasjon MED, har formen:
Hvor b- en konstant verdi for et gitt "adsorbent - adsorbat"-par (forholdet mellom hastighetskonstantene for desorpsjon og adsorpsjon), numerisk lik adsorbatkonsentrasjonen der halvparten av de aktive sentrene er okkupert.

Langmuirs adsorpsjonsisotermgraf er vist i figur 2. Konstanten b bestemme grafisk ved å tegne en tangent til adsorpsjonsisotermen ved punktet MED= 0. Ved beskrivelse av gassadsorpsjonsprosessen i ligningen kan konsentrasjonen erstattes med en proporsjonal verdi av partialtrykket. Teori om monomolekylær adsorpsjon I. Langmuir anvendelig for å beskrive prosessene for adsorpsjon av gasser og oppløste stoffer ved lave trykk (konsentrasjoner) av adsorbatet.
Polyanis teori om polymolekylær adsorpsjon beskriver s-formede adsorpsjonsisotermer, hvis form indikerer mulig interaksjon av adsorberte molekyler med adsorbatet.
1. Adsorpsjon er forårsaket av fysiske krefter.
2. Overflaten av adsorbenten er homogen, det er ingen aktive sentre; adsorpsjonskrefter danner et kontinuerlig kraftfelt nær overflaten av adsorbenten.
3. Adsorpsjonskrefter virker i en avstand større enn størrelsen på adsorbatmolekylet, det vil si at det er et visst adsorpsjonsvolum ved overflaten av adsorbenten, som fylles med adsorbatmolekyler under adsorpsjonen.
4. Tiltrekningen av et adsorbatmolekyl av overflaten av adsorbenten er ikke avhengig av tilstedeværelsen av andre molekyler i adsorpsjonsvolumet, som et resultat av hvilket polymolekylær adsorpsjon er mulig.
5. Adsorpsjonskrefter er ikke avhengig av temperatur, og derfor endres ikke adsorpsjonsvolumet med en endring i temperaturen.
Freundlich ligning. Overflaten til adsorbenten er heterogen, interaksjon skjer mellom de adsorberte partiklene, og de aktive sentrene er ikke helt uavhengige av hverandre. G. Freundlich antydet at antall mol adsorbert gass eller oppløst stoff per masseenhet av adsorbenten (den såkalte spesifikke adsorpsjonen X/m), må være proporsjonal med likevektstrykket (for gass) eller likevektskonsentrasjonen (for stoffer adsorbert fra løsning) av adsorbenten, hevet til en viss styrke, som alltid er mindre enn enhet:
x / m = aP n ; x / m = aC n.
Eksponenter n og proporsjonalitetsfaktor EN bestemt eksperimentelt.
37. Termodynamikk av adsorpsjonsprosessen. Gibbs adsorpsjonsligning
For å studere fenomenet adsorpsjon ved "løsning-gass"-grensesnittet, er det nødvendig å etablere en forbindelse mellom overskuddet av det adsorberte stoffet i laget på overflaten ( G), konsentrasjon av overflateaktive stoffer i løsning ( Med) og overflatespenning ( σ ) ved fasegrensen "løsning - gass". Det er mer hensiktsmessig å vurdere fenomenene fra et termodynamisk synspunkt og relatere adsorpsjonen av et oppløst stoff til en endring i den frie energien til overflaten eller dens overflatespenning. Denne forbindelsen ble opprettet W. Gibbs V 1876, som ble navngitt "Gibbs adsorpsjonsligning":
G = – Med / RT x dσ/dc.
Du kan fortsatt forestille deg Gibbs ligning, basert på termodynamikk, ved bruk av isobarisk-isotermisk potensial G, kjemiske potensialer μ 1 Og μ2, og bruker også n 1 Og n 2 antall mol komponenter. Etter å ha analysert det med hensyn til entropi S, volum V og trykk P, kan vi skrive følgende ligning:
dG=– SDT+VdP+σds+ μ 1 dn 1 + μ 2 dn 2.
La oss likestille det til null, og tatt i betraktning konstant temperatur og trykk, forenkles det til en ligning av formen:
SD σ + n 1 d μ 1 + n 2 d μ 1 = 0.
Tatt i betraktning at for fortynnede løsninger er det kjemiske potensialet til den andre komponenten uttrykt som følger:
μ 2 = μ 2 0 +RT ln c,
og gitt at temperaturen er konstant
dμ 2 =RTdnc,
erstatte denne ligningen i
![]()
får vi den ønskede Gibbs-adsorpsjonsligningen. Basert på ligningen kan man se at hvis overflatespenning σ øker med konsentrasjonen Med, da er konsentrasjonen av det oppløste stoffet på overflatelaget mindre enn i hoveddelen av løsningen (såkalt negativ adsorpsjon), og hvis overflatespenningen σ avtar med økende konsentrasjon Med, da er konsentrasjonen i laget større enn i volumet (positiv adsorpsjon), og til slutt, hvis σ er ikke avhengig av Med, da er konsentrasjonen av stoffet i laget på overflaten og i volumet den samme. Gibbs-ligningen ble utledet ved bruk av termodynamikk. I praksis er det vanskelig å verifisere denne ligningen, noe som skyldes vanskeligheten med å bestemme konsentrasjonen av det oppløste stoffet i lagoverflaten. Erfaren måte B. McBen fant at et veldig tynt lag med væske ble avskåret fra overflaten av løsningen ved hjelp av enheten. Ytterligere bestemmelse av alle parametere i Gibbs-ligningen viste at de eksperimentelt funnet adsorpsjonsverdiene, innenfor den eksperimentelle feilen, falt sammen med verdiene beregnet ved hjelp av Gibbs-ligningen. På grunn av homogeniteten og glattheten til overflaten til enhver væske, er de vanlige konseptene for aktive sentre helt uanvendelige når man studerer adsorpsjon på overflaten. Ved en kritisk temperatur forsvinner forskjellen mellom de tilstøtende fasene, og overflatespenningen blir som regel lik null. Adsorpsjon av gasser og damper har en så bred praktisk anvendelse at man i litteraturen, spesielt i teknisk litteratur, kan finne dette konseptet, som kun brukes i forhold til prosesser på overflaten av faste stoffer.
Dette konseptet, i likhet med de mest generelle adsorpsjonslovene, som Gibbs-ligningen vurdert, er anvendelig for alle fasegrenser. Ved å bruke Gibbs-ligningen og alle bestemmelsene som følger av den, etter å ha bestemt verdien av Γ, er det mulig å konstruere en adsorpsjonsisoterm.
38. Egenskaper ved adsorpsjon på mikroporøse materialer. Polyanys potensielle teori. Adsorpsjonspotensial
Polyanis teori vurderer ikke-lokalisert fysisk adsorpsjon, som er direkte forårsaket av van der Waals-krefter mellom adsorbenten og adsorbatet (dette kan betraktes som den første posisjonen). Den andre posisjonen til denne teorien er ideen om et kraft (eller potensielt) felt av adsorbenten, som strekker seg over en betydelig avstand fra overflaten; adsorpsjonslaget som vises i dette feltet er polymolekylært. Hvis vi vurderer adsorpsjonen av gasser, avtar tettheten til dette laget langs en viss normal fra overflaten. Hvis vi vurderer dampadsorpsjon, dannes et flytende lag av en viss tykkelse på overflaten. Feltet i Polyanis teori betraktes som en serie ekvipotensialflater, hver overflate tilsvarer en viss potensiell verdi ε , og hver påfølgende overflate vil være mindre enn den forrige. Hver slik overflate i rommet kutter ut lag med et visst volum, betegnet som v i. Oppgaven til Polyanyis teori er å finne overgangen fra de vanlige koordinatene til isotermen ( x, s) til feltparametere εi Og v i, med ytterligere etablering av forbindelsen mellom disse grunnleggende parameterne. Den første delen av problemet, som Polyany la ned, er ganske kompleks, og kan i mange tilfeller ikke ha klare løsninger, men når det gjelder dampadsorpsjon, er denne delen av problemet løst i en første tilnærming veldig enkelt. For et væskeadsorpsjonslag vil den fylte delen av volumet være lik:
v i = x(M/d),
Hvor d– tetthet av stoffet i flytende tilstand.
I sin teori introduserer M. Polyany en annen bestemmelse om fraværet av den såkalte. feltscreening under adsorpsjon, verdi ε i denne teorien er rom en konstant verdi (noe sånt som gravitasjonspotensialet) uavhengig av om visse adsorbatmolekyler eksisterer mellom et gitt punkt og en fast overflate eller om alt rom er ledig. Polyani introduserer konseptet adsorpsjonspotensial ε , som representerer det isotermiske arbeidet med kompresjon av damp når den overføres fra likevektstrykk R i bulkfasen bort fra overflaten inn i området av overflatelaget med mettet damptrykk p 0 da vil uttrykket for å bestemme potensialet se slik ut:
ε = RT ln R 0 / R.
Ved å bruke denne ligningen kan du gå fra koordinatene x, p til koordinatene ε Og v og få en kurve, som kalles "karakteristikk". Polyani oppdaget i sine eksperimenter at slike kurver, konstruert fra eksperimentelle data fra de oppnådde isotermene, har følgende egenskap: de er invariante i forhold til T, eller med andre ord, alle kurver av denne typen kan passe på en kurve ε −ε .
M. Polyany aksepterte denne stillingen som et postulat, dvs.:
Denne Polyani-egenskapen er av stor praktisk betydning; den kan konstruere en familie av isotermer fra en eksperimentell adsorpsjonsisoterm.
Polanyis teori gir ikke et analytisk uttrykk for isotermen eller potensialvolumfunksjonen, men den lar en beregne koordinaten for en gitt temperatur hvis minst en isoterm er kjent. Dette resultatet er svært viktig for teknologiske beregninger, fordi for like gasser på samme adsorbent kan adsorpsjonskurvene ligge nær hverandre og kan kombineres i mange tilfeller.
39. Karakteristisk adsorpsjonskurve. Temperaturinvarians og affinitet til karakteristiske kurver
Kraftfeltet som oppstår ved overflaten av adsorbenten kan på mange måter ligne på gravitasjonsfeltet. I adsorpsjonsfeltet kan man tenke seg potensielle overflater, dvs. overflater som er preget av det samme adsorpsjonspotensialet. Under begrepet adsorpsjonspotensial θ skal ikke forstås som noe annet enn arbeidet som gjøres mot adsorpsjonskrefter når man flytter 1 mol adsorbent fra et bestemt punkt i feltet til en bestemt gassfase. Det maksimale adsorpsjonspotensialet vil eksistere ved grensen "adsorbent – adsorpsjonsvolum". Men ved "volum - gassfase"-grensen (det er her virkningen av adsorpsjonskrefter slutter), bør adsorpsjonspotensialet være lik null. Endringen i adsorpsjonspotensial med endring i adsorpsjonsvolumet kan representeres i form av kurver. Dette ble gjort for første gang av M. Polyani. Disse typer kurver er ikke avhengige av temperatur og kan være karakteristiske for hver spesifikke adsorbent; disse typer kurver kalles vanligvis karakteristiske adsorpsjonskurver. Teorien om polymolekylær adsorpsjon antar at gassligningen av tilstand er anvendelig for adsorpsjonsvolumet. Følgelig ligner isotermene som karakteriserer avhengigheten av tettheten til adsorbenten av volumet for forskjellige temperaturer isotermene for avhengigheten av trykk på volum. Ved lave temperaturer kan overflateadsorpsjonskrefter føre til at damp kondenserer til en væske med en viss tetthet. Ved temperaturer lavere enn kritisk, under kondensering, vil hele adsorpsjonsvolumet være fylt med væske. I dette tilfellet vil adsorpsjonskurven løpe nesten parallelt med abscisseaksen, noe som er assosiert med væskens lave komprimerbarhet. Da synker adsorpsjonskurven ved "volum - gassfase"-grensen kraftig ned, og følgelig når tettheten til adsorbenten en viss tetthet av gassfasen. Ved temperaturer høyere enn den kritiske temperaturen kan adsorptivet oppføre seg som en ideell gass, og grafen vil uttrykkes som en ideell gassisoterm, forutsatt at pV = RT. Under slike forhold vil den adsorberte gassen ha en maksimal tetthet ved selve overflaten av adsorbenten og en minimumsdensitet i umiddelbar nærhet av gassfasen. Dessuten, i dette tilfellet, er det viktig å merke seg at tettheten til adsorbenten i adsorpsjonslaget ingen steder når tettheten til selve væsken. Og hvis temperaturen er veldig nær kritisk, vil tetthetens avhengighet av volum uttrykkes ved en kurve nær isotermen, som er beskrevet van der Waals ligning. I denne situasjonen vil en del av det adsorberte stoffet være i det adsorberte volumet i flytende tilstand, og en del av det adsorberte stoffet vil være i gassform. Da vil kurven minke kraftigst i snittet som tilsvarer overgangen fra væske til gass. Hvis du konstruerer en karakteristisk kurve fra den eksperimentelle adsorpsjonsisotermen til et av adsorptivene, og kjenner de tilsvarende affinitetskoeffisientene for et annet adsorptivt, kan du finne adsorpsjonsisotermen og konstruere den for et annet adsorptiv. Potensialteorien om adsorpsjon gjør det mulig å beregne forskjellige adsorpsjonsisotermer av forskjellige damper på samme adsorbent, ved å bruke en karakteristisk kurve som er hentet fra adsorpsjonsisotermen til en damp, siden forholdet mellom adsorpsjonspotensialet ikke er avhengig av adsorpsjonsvolumene .
Affinitet(fra latin affinis - "relatert") - affinitetskromatografi. Metoden for rensing og separering av proteiner er basert på deres selektive interaksjon med en ligand kovalent bundet til en inert bærer (affinitetskromatografi). Måling av affiniteten til et giftstoff for en reseptor er i hovedsak en eksperimentell studie av forholdet mellom mengden av et stoff tilsatt til inkubasjonsmediet og mengden av giftstoff-reseptorkomplekset som dannes som et resultat av interaksjon.
Adsorpsjon skjer i grensesnittet. Derfor er det rimelig å betrakte den termodynamiske beskrivelsen av overflatefenomener som et spesielt tilfelle av termodynamikken til heterogene systemer.
Ris. 3.4. Gibbs-adsorpsjon: 1- tofaset sammenligningssystem, 2- ekte tofasesystem med et uensartet område
I termodynamikken til heterogene systemer brukes det additivitetsprinsippet som er som følger: alle ekstensive egenskaper til et heterogent system er lik summen av de tilsvarende ekstensive egenskapene som fasene ville hatt før de ble brakt i kontakt. La oss betegne fasene med α og β (fig. 4). For et ideelt system, slik at egenskapene til fasene nær grensesnittet faller sammen med deres bulkegenskaper, er følgende relasjoner gyldige for den indre energien U, volum V, masse (antall mol) n, entropi S etter etablering av likevekt i et heterogent system:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
Dette forutsetter at temperaturen og trykket i begge faser er det samme.
For virkelige heterogene systemer gir overgangsregionen ved grensen til to faser et ekstra bidrag til systemets omfattende egenskaper. Dersom overflatefenomener oppstår, bør man ta hensyn til forskjellen mellom de omfattende egenskapene til et reelt heterogent system og de omfattende egenskapene til et modellsystem der overflatefenomener er fraværende. Et slikt system kalles et sammenligningssystem. Sammenligningssystemet har de samme intensive parameterne (T, P, C i ...) og samme volum V som det virkelige systemet (fig. 4).
Fra et termodynamisk synspunkt forstås adsorpsjonsverdien G som den overskytende mengde stoff n s, uttrykt i mol eller gram, som et reelt heterogent system har sammenlignet med referansesystemet, relatert til grensesnittområdet eller til overflatearealet. av adsorbenten A. Det antas at sammenligningssystemet har de samme intensive parameterne (T, P, C i), og samme volum (V = V α + V β) som det virkelige systemet (fig. 4) .
Г = (n - n α - n β)/A = n s/A 3.11
Overskytende termodynamiske funksjoner i overgangsregionen til et reelt system (vi betegner dem med indeksen s) kan skrives som
U s = U - U α - U β , n s = n - n α - n β , S s = S - S α - S β etc.
Eksperimentelle målinger av adsorpsjon gir alltid adsorpsjon nøyaktig som et overskudd av komponenten i det reelle systemet sammenlignet med det valgte referansesystemet. For eksempel, når adsorpsjon av gass på en fast adsorbent eller når adsorpsjon av komponenter på en fast fase, for å finne adsorpsjonsverdier, bestemme endringen i de initiale konsentrasjonene av adsorbatet etter kontakten av fasene α og β
n i s = V(Ci o - C i),
Hvor C i o– innledende konsentrasjon av den i-te komponenten, C i– konsentrasjon av den i-te komponenten etter etablering av likevekt mellom kontaktfasene. Det antas at volumet V endres ikke. Imidlertid konsentrasjonen Jeg komponent C i, oppnådd eksperimentelt, bestemmes i volum V' over fasegrensesnittet uten å ta hensyn til volumet til det inhomogene området av overgangslaget V α ved grensesnittet der konsentrasjonen er C i a. På grunn av eksistensen av en ikke-uniform region i et reelt system, kan det totale volumet av systemet representeres som V = V' + V α. All mengde Jeg-te komponenten C i o vil bli fordelt mellom disse to bindene:
V C i o = V’ C i + V α C i α ,
og antall mol av komponenten Jeg, adsorbert på grensesnittet, vil være lik
n i s = (V’C i + V α C i α) – (V’ + V α)C i = V α (C i α – C i) 3.12
De. eksperimentelt bestemt adsorpsjon er overskuddet av den i-te komponenten i volumet V α sammenlignet med mengden av denne komponenten i samme volum langt fra fasegrensesnittet. Denne typen adsorpsjon kalles Gibbs adsorpsjon. .
V α C i α kalt fullstendig innhold Jeg- komponent i adsorpsjonslaget. I området med svært lave konsentrasjoner C i i volum V' endring V α C i ligning (3.2) kan neglisjeres og den målte verdien kan vurderes V α C i α fullt innhold Jeg- komponent i adsorpsjonslaget, for eksempel under gassadsorpsjon på en fast adsorbent ved lavt trykk.
Adsorpsjon som en spontan konsentrasjon av molekyler på en overflate er ledsaget av en reduksjon i systemets entropi. Siden kriteriet for prosessens spontanitet er
∆H - T · ∆S = ∆G< 0,
da er adsorpsjon kun mulig ved ∆H< 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. Når temperaturen øker, skifter likevekten mot den endoterme prosessen, dvs. desorpsjon.
Adsorpsjon på fast overflate
1. Monomolekylær adsorpsjon.
I følge Langmuirs teori samhandler adsorberende molekyler med overflaten av adsorbenten, og danner til slutt et monomolekylært lag. I dette tilfellet, graden av fylling () av overflaten med det adsorberte stoffet under adsorpsjon fra gassfasen
fra væske
hvor K er likevektskonstanten (adsorpsjonskonstanten);
p er partialtrykket til den adsorberte gassen;
c er konsentrasjonen av det adsorberte stoffet.
Avhengigheten av β av p (eller c) er presentert av grafen (adsorpsjonsisoterm, T = const) i fig. 1.3.

Ris. 1.3. Grad av overflatefylling med adsorbert stoff
Ved lave konsentrasjoner og partialtrykk er adsorpsjonen proporsjonal med konsentrasjonen eller partialtrykket:
R<< 1, β ≈ К· r ilis<< 1, β ≈ К· s, dvs. den innledende delen av isotermen er tilnærmet lineær, og tan α = K (tg α bestemmes av stigningstallet på kurven ved p (eller c) → 0: eller ).
If er antall mol adsorbert stoff per 1 g adsorbent; - maksimalt mulig antall mol adsorbert stoff per 1 g adsorbent ("monolagskapasitet"), deretter
Sette inn β i ligning (1.3) (for tilfellet med adsorpsjon fra gassfasen, konsentrasjonen Med i ligninger bør erstattes av trykk R), vi får:
 (1.6)
(1.6)
Siden og K i et gitt adsorbent-adsorbentpar er konstanter (ved T=const), så ved avhengighet kan man finne TIL(Fig. 1.4).

Ris. 1.4. Grafisk løsning av adsorpsjonsligningen
oppnådd ved å ekstrapolere den eksperimentelle lineære avhengigheten til () = 0; og siden , da .
Verdien kan brukes til å bestemme det spesifikke overflatearealet til adsorbenten UD (i m 2 per 1 g adsorbent), hvis arealet ω okkupert på overflaten av ett molekyl av adsorbenten er kjent (bestemt ut fra størrelsen på molekylet):
UD = · ω · Na, (1,7)
hvor Na er Avogadros tall (Na = 6,02 10 23).
I sin tur kan den kjente verdien av UD brukes til å beregne ω av ethvert stoff basert på dets adsorpsjon på en gitt adsorbent.
2. Polymolekylær adsorpsjon.
Ligning (1.5) beskriver en kurve med metning, dvs. på
p (eller c) → ∞ har en tendens til grenseverdien lik (fig. 1.5,a).

Fig.1.5. Adsorpsjonsisotermer:
a - adsorpsjon med metning; b – polymolekylær adsorpsjon
I noen tilfeller ser imidlertid adsorpsjonsisotermer ut som de som er vist i fig. 1,5, b, dvs. når ikke grensen selv ved høy p (eller c).
Avhengigheter av typen vist i fig. 1,5,b tilsvarer polymolekylær adsorpsjon. Som regel er slike isotermer karakteristiske for stoffer med sterke intermolekylære interaksjoner (for eksempel vann). Når adsorpsjonssentrene på overflaten av adsorbenten er opptatt (det monomolekylære laget er mettet), skjer "landingen" av de neste adsorbatmolekylene på grunn av intermolekylære interaksjoner med allerede adsorberte molekyler (fig. 1.6). Varmen fra slik adsorpsjon er nær i absolutt verdi, men motsatt i fortegn til fordampningsvarmen til den tilsvarende væsken (tenk på hvorfor).

Fig.1.6. Adsorpsjonsskjema:
a - monomolekylær adsorpsjon; b - polymolekylær adsorpsjon
Når vi kommer nærmere R til det mettede damptrykket til det adsorberte stoffet begynner det å kondensere på overflaten av adsorbenten, som et resultat vokser det raskt med økende R.
Ved interaksjon mellom to atomer:
U – interaksjonsenergi;
U = U FORR. + U RETUR
 - Lennard-Jones ligning
, c, b, m = konst
- Lennard-Jones ligning
, c, b, m = konst
I tilfeller av interaksjon av atomer med en fast overflate, er det nødvendig å oppsummere alle interaksjoner.
x – avstand til overflaten
r – virkningsradius for tiltrekningskrefter
dV – volum
n – antall overflatemolekyler
U ADS. – energi av adsorpsjonsinteraksjon



Ved adsorpsjon øker tiltrekningen. Og når det gjelder ikke-polar-ikke-polar interaksjon, er adsorpsjonen hovedsakelig lokalisert i fordypninger.
Elektrostatisk interaksjon.
Polar adsorbent – ikke-polart adsorbat
Ikke-polar adsorbent – polar adsorbat
Polar adsorbent – polar adsorbat.
M  Adsorbatmolekylet er representert som en dipol, og adsorbenten er representert som en leder der adsorbatmolekylet induserer et dipolspeil symmetrisk i forhold til det gitte.
Adsorbatmolekylet er representert som en dipol, og adsorbenten er representert som en leder der adsorbatmolekylet induserer et dipolspeil symmetrisk i forhold til det gitte.
X – avstand til midten
Når du samhandler, oppstår potensialet:
 ,
,
 - dipolmoment.
- dipolmoment.
Potensialet har en tendens til å ta på seg maksimalverdien, dvs. dipoler har en tendens til å orientere seg vinkelrett på overflaten.
Siden en økning i temperatur fremmer veksten av Brownsk bevegelse, fører det til hemming av adsorpsjonsprosessen.
Ved elektrostatisk interaksjon er adsorbatet hovedsakelig lokalisert på fremspringene.
Fundamental adsorpsjonsligning.
Ved adsorpsjon skjer det en omfordeling av komponenten, som betyr at det kjemiske potensialet endres. Adsorpsjonsprosessen kan betraktes som overgangen av overflateenergi til kjemisk energi.
Lagvolum = 0, deretter den generaliserte ligningen for termodynamikkens I- og II-lover:
T = const; (1) = (2) => 

For et to-komponent system:
,
 ,
,
 =>
=>
=>
 - Gibbs adsorpsjonsligning
.
- Gibbs adsorpsjonsligning
.
For TV-adsorpsjon. kropp - gass: , 
 ,
,

 - isoterm
- isoterm
 - isobar
- isobar
 - isopycnal
- isopycnal
 - isoster
- isoster
Isoterm, isopycne, isostere er relatert til hverandre.
Fordi adsorpsjonsfunksjon 



Henry isoterm Langmuir isoterm



Termodynamikk. Adsorpsjon.
For kondensert materiale:
 ,
,
 ,
,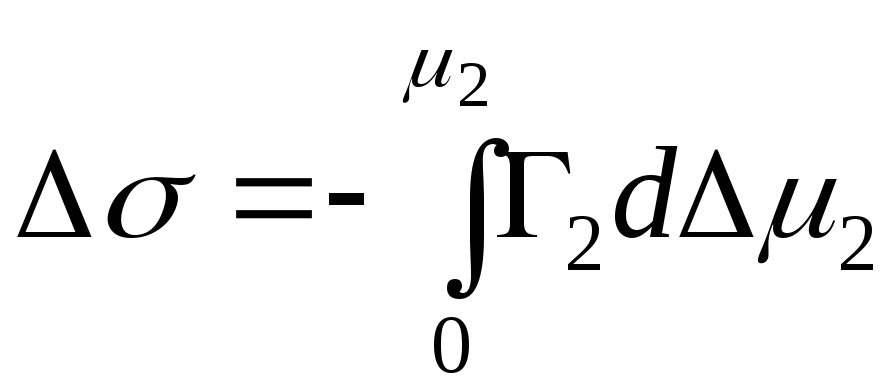
 - integrert endring i Gibbs energi
.
- integrert endring i Gibbs energi
.

P – trykk over en buet overflate, Р S – trykk over en flat overflate
 - adsorpsjonspotensial
- adsorpsjonspotensial
Differensiell endring i entrapi 
 , Г = konst
, Г = konst
- differensiell entropi endring
- differensiell entalpi av adsorpsjon
 - isosterisk adsorpsjonsvarme
- isosterisk adsorpsjonsvarme
 - kondensasjonsvarme
- kondensasjonsvarme
 - netto adsorpsjonsvarme
- netto adsorpsjonsvarme

 ,
,




Qa – integrert adsorpsjonsvarme, 
Qra – integrert netto adsorpsjonsvarme,

Henrys ligning
Studiet av adsorpsjon er komplisert av overflatens heterogenitet, så de enkleste lovene oppnås for homogene overflater.
La oss vurdere samspillet mellom gasser og en fast overflate, når en gass går over fra en likevektstilstand i volumet til en likevektstilstand på overflaten. Dette tilfellet er analogt med likevekten til gasser i et gravitasjonsfelt.
 ,
,
 ,
=>
,
=> -Henrys ligning
-Henrys ligning
 - fordelingskoeffisient
- fordelingskoeffisient
Under adsorpsjonsprosessen skjer det en endring i kjemiske potensialer.
For bulkfasen: 
For gass på overflaten: 
I en tilstand av balanse  , dvs.
, dvs.

I Henrys ligning er konstanten ikke avhengig av konsentrasjon
Henrys ligning er gyldig i området med lave trykk og konsentrasjoner. Når konsentrasjonen øker, er 2 typer avvik fra Henrys lov mulig:
1 – positive avvik, D minker, A minker
2 – negative avvik, D – øker, A – øker.
Typen av avvik bestemmes av overvekten av en eller annen type adsorbent-adsorbat-interaksjon.
Med sterk adhesiv interaksjon øker aktivitetskoeffisientene - et positivt avvik. Ved sammenhengende interaksjoner observeres negative avvik.
Monomolekylær adsorpsjon.
Langmuir isoterm.
De enkleste mønstrene ble oppnådd i Henrys teori. Langmuir foreslo en teori der adsorpsjon anses som en kvasikjemisk reaksjon. Hvori:
Overflaten er energisk homogen.
Adsorpsjon er lokalisert, hvert adsorpsjonssenter samhandler med ett adsorbatmolekyl.
Adsorbatmolekyler interagerer ikke med hverandre.
Monolag adsorpsjon.

 - overflate,
- overflate,  - adsorbat,
- adsorbat,  - adsorpsjonskompleks.
- adsorpsjonskompleks.
 , deretter konsentrasjonen av adsorpsjonssteder:
, deretter konsentrasjonen av adsorpsjonssteder:  ,
, - begrenser adsorpsjon.
- begrenser adsorpsjon.
 , da er reaksjonskonstanten:
, da er reaksjonskonstanten: 

 - Langmuir-ligningen.
- Langmuir-ligningen.
Avhengighet av adsorpsjon av konsentrasjon
1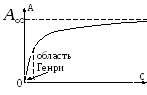 )
)
 ,
,

2) område med høye konsentrasjoner
 - begrensende adsorpsjon, dannelse av et monomolekylært lag
- begrensende adsorpsjon, dannelse av et monomolekylært lag
For Gibbs energi: .
g er entropifaktoren.
Når det gjelder Henry-isotermen, karakteriserer Gibbs-energien overgangen til adsorbatet fra standardtilstanden i bulk til standardtilstanden på overflaten. I tilfellet med Langmuir-isotermen  karakteriserer graden av affinitet mellom adsorbenten og adsorbatet.
karakteriserer graden av affinitet mellom adsorbenten og adsorbatet.
 funnet fra van't Hoff isobar.
funnet fra van't Hoff isobar.

 , Deretter
, Deretter  , herfra
, herfra  .
.

 - grad av overflatefylling.
- grad av overflatefylling.




 - antall ledige plasser,
- antall ledige plasser,  - antall besatte plasser.
- antall besatte plasser.
 ,
,

De. i området med høye konsentrasjoner er antallet frie steder omvendt proporsjonalt med mengden adsorbat.
Adsorpsjon av en blanding av gasser på en homogen overflate.
I dette tilfellet betraktes adsorpsjonsprosessen som to parallelle reaksjoner.
 (1)
(1)

 (2)
(2)


Adsorpsjon av en blanding av gasser på en ujevn overflate.
Ved ujevn overflate kan man ikke begrense seg til gjennomsnittlige fyllinger.
Som et resultat av konkurranse er lokalisering av forskjellige adsorbater mulig i områder av forskjellige typer.
I dette tilfellet forholdet  .
.
 ,
,
 - mettet damptrykk av adsorbatet.
- mettet damptrykk av adsorbatet.
 ,
,
 - adsorpsjonsvarme.
- adsorpsjonsvarme.
"+" - symbatavhengighet, "-" - antibatavhengighet, "N" - ingen korrelasjon.
"+" - adsorpsjon fortsetter i henhold til samme mekanisme. I de mest energimessig gunstige områdene blir gass med høy affinitet til overflaten hovedsakelig adsorbert.
"-" - adsorpsjon skjer gjennom ulike mekanismer og inntil et visst tidspunkt er det ingen konkurranse om overflaten.
Monomolekylær adsorpsjon realiseres hovedsakelig under fysisk adsorpsjon av gasser ved lave verdier s, så vel som ved væske/gass-grensesnittet.
Polymolekylær adsorpsjon.
BET teori(Brunauer, Emmett, Teller).
I tilfellet hvor dannelsen av et monolag ikke er nok til å kompensere for overflateenergien, er adsorpsjonen polymolekylær og kan betraktes som et resultat av tvungen kondensering under påvirkning av overflatekrefter.
Viktige punkter:
Når et adsorbatmolekyl treffer et okkupert sted, dannes et multippelsett.
Når vi kommer nærmere s Til s s antall gratis adsorpsjonssteder reduseres. Til å begynne med øker antall plasser besatt av singler, doubler osv. for deretter å reduseres. i sett.
På s =s s adsorpsjon blir til kondens.
Det er ingen horisontale interaksjoner.
For det første laget er Langmuir-isotermen oppfylt.
Overflaten betraktes som et sett med adsorpsjonssteder. Betingelsen for dynamisk likevekt er gyldig: kondenseringshastigheten på frie steder er lik fordampningshastigheten fra okkuperte steder.
a er kondensasjonskoeffisienten (andelen av molekyler som kondenseres på overflaten);
 ,
,
Zm – maksimalt antall ledige plasser.
 - frekvens av atomvibrasjoner i retning vinkelrett på overflaten.
- frekvens av atomvibrasjoner i retning vinkelrett på overflaten.
For det første laget, dynamiske likevektsforhold:



 , Deretter
, Deretter
 - Langmuir-ligningen.
- Langmuir-ligningen.
For det andre laget vil det være sant: 
For det i-te laget: 
For enkelhets skyld antas det at a og ν er like for alle lag bortsett fra det første. For alle lag unntatt det første er adsorpsjonsvarmen konstant. For det siste laget er adsorpsjonsvarmen lik kondensasjonsvarmen. Som et resultat ble ligningen oppnådd
 (*)
(*)
C- konstant,
Når det gjelder BET-teori, konstanten MED karakteriserer Gibbs-energien til ren adsorpsjon. Ligningen inneholder bare en konstant, og denne ligningen er også veldig viktig for å bestemme det spesifikke overflatearealet til adsorbenten.
Siden varme frigjøres som følge av adsorpsjon, bestemmes spesifikke overflatearealer ved lave temperaturer.
 ????????????
????????????


Den største ulempen med teorien– neglisjering av horisontale interaksjoner til fordel for vertikale.
Ligningen holder i området  fra 0,05 til 0,3.
fra 0,05 til 0,3.
Hvor  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 – interaksjonen adsorbat – adsorbat påvirkes.
> 0,3 – interaksjonen adsorbat – adsorbat påvirkes.
Redegjørelse for adsorbat-adsorbat interaksjoner.
Interaksjoner oppstår når forgrenede molekyler eller molekyler adsorberes på en ikke-polar overflate. I stand til å danne medarbeidere. I dette tilfellet endres formen på adsorpsjonsisotermene.
EN  adsorbenten er ikke polar.
adsorbenten er ikke polar.
Graf 1 tilsvarer svake adsorbat-adsorbat-interaksjoner og sterke adsorbat-adsorbent-interaksjoner.
Graf 2 tilsvarer sterke adsorbat-adsorbat og sterke adsorbat-adsorbent interaksjoner.
Graf 3 tilsvarer sterk adsorbat-adsorbat-interaksjon og svak adsorbat-adsorbent-interaksjon.
 ,
,

Ved interaksjon mellom adsorbatmolekyler er det nødvendig å ta hensyn til endringer i aktivitetskoeffisienter. Og denne ligningen er skrevet som:
 - Frunkin, Fowler, Guggenheim-ligning.
- Frunkin, Fowler, Guggenheim-ligning.
k– tiltrekningskonstant.
Polyanys potensielle teori.
Denne teorien utleder ikke noen form for adsorpsjonsisoterm, men gjør det mulig å beregne isotermer ved en annen temperatur.
Adsorpsjon- dette er resultatet av tiltrekningen av adsorbatet til overflaten av adsorbenten på grunn av virkningen av adsorpsjonspotensialet, som ikke er avhengig av tilstedeværelsen av andre molekyler og avhenger av avstanden mellom overflaten og adsorbatmolekylet.
 ,
,
 - adsorpsjonspotensial.
- adsorpsjonspotensial.
Siden overflaten er ujevn, erstattes avstanden med adsorpsjonsvolumet  .Adsorpsjonsvolum er volumet innelukket mellom overflaten og punktet som tilsvarer en gitt verdi
.Adsorpsjonsvolum er volumet innelukket mellom overflaten og punktet som tilsvarer en gitt verdi  .
.
Adsorpsjonspotensial er arbeidet med å overføre 1 mol adsorbat utenfor et gitt adsorpsjonsvolum til et gitt punkt i adsorpsjonsvolumet (eller arbeidet med å overføre 1 mol mettet damp av et adsorbat som er i likevekt med et flytende adsorbat i fravær av en adsorbent inn i en dampfase i likevekt med adsorbenten).

Karakteristisk kurve
 - adsorpsjonspotensial,
- adsorpsjonspotensial, 
For en gitt adsorbent og forskjellige adsorbater er følgende sant: 
For ulike typer adsorbater  ,
,
Hvor  potensialer for adsorpsjonsisotermer ved relative trykk
potensialer for adsorpsjonsisotermer ved relative trykk  for adsorbat 1 og for adsorbat 2. Dette forholdet er en konstant verdi.
for adsorbat 1 og for adsorbat 2. Dette forholdet er en konstant verdi.
 - affinitetskoeffisient
- affinitetskoeffisient
Teori om kapillær kondensasjon.
Forløpet av adsorpsjonsprosessen avhenger i stor grad av strukturen til den porøse kroppen.
|
Mikroporøs | |
|
Overgangsporøs | |
|
Makroporøs |
Når det gjelder mikroporøse sorbenter, overlapper feltene med adsorpsjonskrefter. Ved makroporøse sorbenter fungerer porene som transportkanaler. Kondensasjonsprosesser er mest betydningsfulle i overgangsvis porøse kropper. Kapillærkondensering begynner ved visse verdier s Og  , når en del av overflateenergien allerede er kompensert. En nødvendig betingelse er at overflaten skal være selvfuktende. Prosessen er beskrevet Thompson-Kelvin-ligningen.
, når en del av overflateenergien allerede er kompensert. En nødvendig betingelse er at overflaten skal være selvfuktende. Prosessen er beskrevet Thompson-Kelvin-ligningen.

 - for fukting er krumningssenteret i gassfasen.
- for fukting er krumningssenteret i gassfasen.
Ved kapillærkondensering har adsorpsjonsisotermen en hysteretisk form. Den nedre grenen tilsvarer adsorpsjonsprosessen, og den øvre grenen tilsvarer desorpsjonsprosessen.
Alle typer porer kan reduseres til tre typer:
|
Konisk |
Sylindrisk med en lukket ende |
Sylindrisk med to åpne ender |
|
Prosessfylling utføres fra bunnen av poren. Adsorpsjonsisotermen og desorpsjonsisotermen faller i dette tilfellet sammen, siden adsorpsjonsprosessen starter fra en kule og desorpsjonsprosessen også begynner med at noen kuler forsvinner.
↓ |
Det er ingen hysterese. Forover og bakover slag er beskrevet av ligningen:
|
Det er ingen bunn noe sted, fyllingen av poren vil gå langs sylinderens vegger.
sylinder: Isotermen vil ha et hysteretisk utseende.
↓ |


 - sfære,
- sfære, ,
,



I  Under fuktige forhold oppstår kondens ved lavere trykk, noe som er energetisk gunstig. Fra desorpsjonsgrenen oppnås porestørrelsesfordelingskurver.
Under fuktige forhold oppstår kondens ved lavere trykk, noe som er energetisk gunstig. Fra desorpsjonsgrenen oppnås porestørrelsesfordelingskurver.
Differensialkurvens maksimum forskyves til venstre i forhold til integralkurvens infleksjonspunkt. Det totale volumet av små porer er lite, men har store overflatearealer. Med økende porestørrelse øker volumet deres som  , og området er som
, og området er som  , på grunn av dette observeres et skift i maksimum av differensialkurven.
, på grunn av dette observeres et skift i maksimum av differensialkurven.
Adsorpsjon ved fast-væske-grensesnittet.
Når det gjelder adsorpsjon ved fast-gass-grensesnittet, forsømte vi en komponent. Ved adsorpsjon ved faststoff-væske-grensesnittet, fortrenger adsorbatet løsemiddelmolekyler fra overflaten av adsorbenten.
 ,
,
Ligningen er riktig:

 ,
,
N 1, N 2 – molfraksjoner av løsemiddel og komponent, N 1 + N 2 = 1, deretter
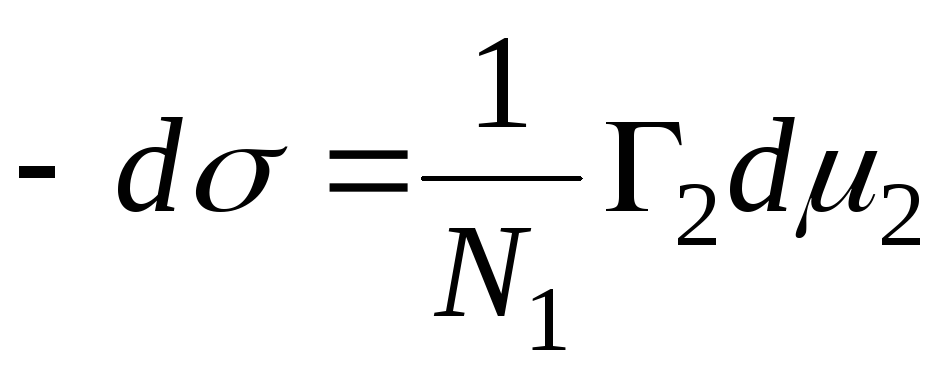 ,
=>
,
=>
 , så er adsorpsjonsligningen for fast-væske-grensesnittet.
, så er adsorpsjonsligningen for fast-væske-grensesnittet.
Adsorpsjon (G) > 0 ved  <
0
<
0
Hvis verdiene  for komponenten og løsningsmidlet er svært forskjellige, i dette tilfellet avhengigheten G fra N har et ekstremum ved verdien N
~ 0,5.
for komponenten og løsningsmidlet er svært forskjellige, i dette tilfellet avhengigheten G fra N har et ekstremum ved verdien N
~ 0,5.
E  hvis
hvis  har nære verdier, i dette tilfellet kan tegnet på adsorpsjon endres. Avhengighet G fra N krysser x-aksen
har nære verdier, i dette tilfellet kan tegnet på adsorpsjon endres. Avhengighet G fra N krysser x-aksen
Funksjon skjæringspunkt G(N) med x-aksen kalles adsorpsjonsazeotrop. Dette betyr at de to komponentene ikke kan separeres på en gitt adsorbent.
Ligning av adsorpsjon isoterm med utvekslingskonstant.
Under adsorpsjon ved faststoff-væske-grensesnittet skjer en konstant omfordeling av komponenter mellom overflaten av adsorbenten og volumet av løsningen.
 - komponenter (- - referer til overflaten)
- komponenter (- - referer til overflaten)

 ,
,
 ,
, .
.
 ,
,


Adsorpsjon ved væske-gass-grensesnittet
R  La oss vurdere endringen i konsentrasjonsprofilen når væske-gass-grensesnittet krysses. La komponent 2 være flyktig.
La oss vurdere endringen i konsentrasjonsprofilen når væske-gass-grensesnittet krysses. La komponent 2 være flyktig.
Cs – konsentrasjon i overflatelaget.
Basert på definisjonen av overflødig adsorpsjon

Hvis komponenten ikke er flyktig, vil adsorpsjonsverdien bli skrevet som følger:
P  ri
ri 

I Eq.  naturen til et stoff er beskrevet av dets derivat
naturen til et stoff er beskrevet av dets derivat  .
.
Overflatespenningsisotermen kan ha formen 1 eller 2:
1 - overflateaktive stoffer
2 - overflateaktive stoffer
Overflateaktivitet g er stoffenes evne til å redusere overflatespenningen i et system.

 - tykkelse på overflatelaget
- tykkelse på overflatelaget
C s– konsentrasjon av komponenten i overflatelaget
MED– volumkonsentrasjon
For en homolog serie er det en regel:
 - Traubo Duclos regel
- Traubo Duclos regel
For en homolog serie ser adsorpsjonsisotermen slik ut:
I stedet for A skriver vi G, siden adsorpsjonen er for stor i overflatelaget.
Overflatespenningsisoterm:

 - overflatespenning av et rent løsemiddel.
- overflatespenning av et rent løsemiddel.
 - fundamental adsorpsjonsligning;
- fundamental adsorpsjonsligning;
 - Langmuir-ligningen.
- Langmuir-ligningen.
La oss løse dem sammen:

- Shishkovsky-ligningen.
B– konstant for den homologe serien.
EN- når du flytter fra en homolog til en annen øker med 3-3,5 ganger 
![]()
1 – område med lave konsentrasjoner
![]()
2 – gjennomsnittlig konsentrasjon
3 – monomolekylært lag
Surfaktanter er difile molekyler, dvs. inkluderer en polar gruppe og et ikke-polart hydrokarbonradikal.
o er den polare delen av molekylet.
| - ikke-polar del av molekylet.
I et polart løsningsmiddel er overflateaktive molekyler orientert på en slik måte at den polare delen av molekylet vender mot løsningsmidlet, og den ikke-polare delen skyves inn i gassfasen.
I Shishkovskys ligning  , den er konstant for den homologiske serien.
, den er konstant for den homologiske serien.
Den overflateaktive effekten begynner å vises med n>5. Ved konsentrasjoner høyere enn konsentrasjonen av det monomolekylære laget, skjer micellisering i overflateaktive løsninger.
Micelle– kalles et aggregat av amfifile overflateaktive molekyler, hvis hydrokarbonradikaler danner en kjerne, og de polare gruppene omdannes til den vandige fasen.
Micelle masse – micelle masse.
H  antall molekyler – aggregeringsnummer.
antall molekyler – aggregeringsnummer.
Sfæriske miceller
Ved micellisering etableres likevekt i løsningen
CMC – kritisk konsentrasjon av micelledannelse.
Siden vi anser micellen som en egen fase:
For en homologisk serie er det en empirisk ligning:
en– energi for oppløsning av den funksjonelle gruppen.
b – økning av adsorpsjonspotensial, adsorpsjonsarbeid per metylenenhet.
– økning av adsorpsjonspotensial, adsorpsjonsarbeid per metylenenhet.
Tilstedeværelsen av en hydrokarbonkjerne i miceller skaper muligheten for at forbindelser som er uløselige i vann kan løses opp i vandige løsninger av overflateaktive stoffer; dette fenomenet kalles solubilisering (det som løses opp er solubilisatet, det overflateaktive midlet er solubiliseringsmiddelet).
Slammet kan være helt upolart, kan inneholde både polare og ikke-polare deler og vil være orientert som et overflateaktivt molekyl.
I alle fall, under solubilisering er det en økning i micellær masse og aggregeringsantall, ikke bare på grunn av inkludering av solubilisat, men også på grunn av en økning i antall overflateaktive molekyler som er nødvendige for å opprettholde en likevektstilstand.
Solubilisering er mer effektiv, jo lavere molekylvekten til solubilisatet er.

 ~ 72 mN\m.
~ 72 mN\m.
 ~ 33 mN\m.
~ 33 mN\m.
Effektiviteten til overflateaktive stoffer avhenger av CMC-verdien.
2D overflatelagstrykk

→ -overflatespenningskrefter.
- todimensjonalt trykk.
Overflatelaget er en kraft lik forskjellen i overflatespenning av en overflateaktivt løsning og et rent løsningsmiddel, rettet mot en ren overflate.
Det etableres en likevekt mellom løsningen og overflatelaget



På  det er et område hvor
det er et område hvor  avhenger lineært av konsentrasjon.
avhenger lineært av konsentrasjon.
G [mol/m2].
 -området okkupert av en mol av et stoff
-området okkupert av en mol av et stoff
Da vil den todimensjonale trykkisotermen ha formen 
 - todimensjonal trykkisoterm.
- todimensjonal trykkisoterm.
Avhengighet  fra S M:
fra S M:

På  - todimensjonalt trykk øker kraftig. På
- todimensjonalt trykk øker kraftig. På  todimensjonal er deformert, noe som forårsaker plutselig vekst
todimensjonal er deformert, noe som forårsaker plutselig vekst  .
.
En film avgrenset av identiske faser på begge sider kalles dobbeltsidig. I slike filmer observeres konstant bevegelse av moderluten.
Filmer mindre enn 5 nm tykke kalles svarte filmer.

Adsorpsjonslag må ha to egenskaper: viskositet og lett bevegelighet, fluiditet og elastisitet.
Marangoni-effekten er selvhelbredende.
Gibbs trekant,  - overtrykk.
- overtrykk.
Filmen har strukket seg og på grunn av at en del av væsken har forlatt, suser de overflateaktive stoffene inn i det frie rommet. Gibbs trekant.
Effekten av kroppens adsorpsjonsstyrke.
Det er alltid et adsorpsjonslag på overflaten av filmen, som da
Langmuir ligning:


 inn i todimensjonalt trykk
inn i todimensjonalt trykk
 - en analog av Shishkovsky-ligningen
- en analog av Shishkovsky-ligningen
Elektrokinetiske fenomener. Elektrisk dobbeltlag (EDL).
Gelemholtz modell. Gouy-Chapman teori.
1808 Fly

U – formet rør, dypp 2 elektroder inn i det. Loven om kommuniserende kar brytes og det oppstår en endring i væskenivået i røret - elektrokinetiske fenomener.
Kinetiske fenomener:
Elektroforese
Elektroosmose
Strømnings (strømnings) potensial
Sedimentasjonspotensial
1 og 2 oppstår når en potensialforskjell påføres; 3 og 4 forårsaker stansing og sedimentering av kolloidale partikler utseendet til en potensialforskjell.
Elektroosmose er bevegelsen av et dispersjonsmedium i forhold til en stasjonær spredt fase under påvirkning av en elektrisk strøm.
Elektroforese – dette er bevegelsen av dispergerte fasepartikler i forhold til et stasjonært dispersjonsmedium under påvirkning av en elektrisk strøm.
P  Årsaken til forekomsten av elektrokinetiske fenomener er romlig separasjon av ladninger og utseendet til et dobbelt elektrisk lag.
Årsaken til forekomsten av elektrokinetiske fenomener er romlig separasjon av ladninger og utseendet til et dobbelt elektrisk lag.
Det elektriske dobbeltlaget er en flat kondensator, en plate er dannet av potensialbestemmende ioner, den andre av motioner. Ionene forurenses på samme måte som potensialbestemmende koioner presses inn i volumet av løsningen. Avstand mellom platene  . Potensialet synker lineært, potensialforskjellen
. Potensialet synker lineært, potensialforskjellen  .
.
En ekstern potensialforskjell forårsaker utseendet til en skjærmodul  er et par krefter per arealenhet som virker langs overflaten til et fast legeme.
er et par krefter per arealenhet som virker langs overflaten til et fast legeme.

I likevekt er skjærmodulen lik den viskøse friksjonsmodulen (  ).
).

I våre forhold  ,
,

 - Gelemholtz-Smalukowski-ligningen
- Gelemholtz-Smalukowski-ligningen
 - lineær hastighet for faseforskyvning.
- lineær hastighet for faseforskyvning.
E– elektrisk feltstyrke.
 - potensialforskjell mellom plater
- potensialforskjell mellom plater
 - elektroforetisk mobilitet [m 2 /(V*s)].
- elektroforetisk mobilitet [m 2 /(V*s)].
Helemholtz-modellen tar ikke hensyn til den termiske bevegelsen til molekyler. I virkeligheten er fordelingen av ioner i dobbeltlaget mer kompleks.
Gui og Chapman identifiserte følgende årsaker til DES:
Overgangen til et ion fra en fase til en annen når likevekt er etablert.
Ionisering av fastfasestoff.
Komplettering av overflaten med ioner tilstede i dispersjonsmediet.
Polarisering fra en ekstern strømkilde.
Det elektriske dobbeltlaget har en uklar eller diffus struktur. Ionene har en tendens til å være jevnt fordelt gjennom det diffuse laget.
Det diffuse laget består av motinoner; lengden på laget bestemmes av deres kinetiske energi. Ved temperaturer som nærmer seg absolutt null er motioner så nærme som mulig potensialbestemmende ioner.
Danyas teori er basert på to ligninger:
Boltzmann-ligningen

 - arbeid mot kreftene til elektrostatisk interaksjon.
- arbeid mot kreftene til elektrostatisk interaksjon.
 - volumetrisk ladningstetthet.
- volumetrisk ladningstetthet.

Poissons ligning 
Siden tykkelsen på EDL er mye mindre enn partikkelstørrelsen og for en flat EDL den deriverte med hensyn til koordinater  Og
Og  er opphevet.
er opphevet. 
For e y at y<<1 функцию можно разложить в ряд Маклорена:

La oss begrense oss til to termer i serien, da:


 - DEL-tykkelse er avstanden som DEL-potensialet avtar i e en gang.
- DEL-tykkelse er avstanden som DEL-potensialet avtar i e en gang.
Jo lavere temperatur, jo mindre  . Ved T→0 – flat DEL. Jo høyere konsentrasjon, jo mer jeg, jo mindre
. Ved T→0 – flat DEL. Jo høyere konsentrasjon, jo mer jeg, jo mindre  .
.





 "–" betyr at potensialet avtar med avstanden. =>
"–" betyr at potensialet avtar med avstanden. =>
=>

 ,
,
 - potensialet synker eksponentielt.
- potensialet synker eksponentielt.
Potensial for overflateladningstetthet: 
Overflateladning er en volumladning med motsatt fortegn, integrert over avstand.




=>


Der potensialet reduseres med 2,7 ganger - 
Dobbeltlags kapasitet 
Ulempen med teorien er at tilstedeværelsen av Helemholtz-laget ikke tas i betraktning, d.v.s. tar ikke hensyn til  , derav feilene ved å bestemme hovedparametrene. Det forklarer heller ikke påvirkningen av ioner av forskjellig natur på tykkelsen av det elektriske dobbeltlaget.
, derav feilene ved å bestemme hovedparametrene. Det forklarer heller ikke påvirkningen av ioner av forskjellig natur på tykkelsen av det elektriske dobbeltlaget.
Sterns teori. Struktur av en kolloidal micelle.
Det elektriske dobbeltlaget består av to deler: tett og diffust. Et tett lag dannes som et resultat av samspillet mellom potensialdannende ioner med spesifikt adsorberte. Disse ionene er som regel delvis eller fullstendig dehydrert og kan ha enten samme eller motsatt ladning til de potensialbestemmende ionene. Det avhenger av forholdet mellom elektrostatisk interaksjonsenergi  og spesifikt adsorpsjonspotensial
og spesifikt adsorpsjonspotensial  . De tette lagionene er fiksert. Den andre delen av ionene ligger i det diffuse laget, disse ionene er frie og kan bevege seg dypere inn i løsningen, dvs. fra et område med høyere konsentrasjon til et område med lavere konsentrasjon. Den totale ladningstettheten består av to deler.
. De tette lagionene er fiksert. Den andre delen av ionene ligger i det diffuse laget, disse ionene er frie og kan bevege seg dypere inn i løsningen, dvs. fra et område med høyere konsentrasjon til et område med lavere konsentrasjon. Den totale ladningstettheten består av to deler.

 -ladning av Helmholtz-laget
-ladning av Helmholtz-laget
 -Diffus lagladning
-Diffus lagladning
Overflaten har et visst antall adsorpsjonssentre, som hver samhandler med ett motion. Konstanten til en slik kvasikjemisk reaksjon er lik:

 , Hvor
, Hvor  - molfraksjon av motioner i løsning
- molfraksjon av motioner i løsning
Helmholtz distribusjon
Potensialet avtar lineært
Gouy potensiell distribusjon. Det er ikke noe tett lag, potensialet synker eksponentielt fra verdien 
Hekkfordeling.
Til å begynne med er potensialreduksjonen lineær og deretter eksponentiell.
Når et elektrisk felt påføres ved elektroforese, er det ikke partikkelen i den faste fasen som beveger seg direkte, men partikkelen i den faste fasen med et lag av ioner som omgir den. DES gjentar formen til den dispergerte fasepartikkelen. Når et potensial påføres, rives en del av det diffuse laget av. Brytelinjen kalles glidende grense.
Potensialet som oppstår ved glidegrensen som følge av separasjon av en del av det diffuse laget kalles elektrokinetisk potensial(Zeta-potensial  ).
).
En dispergert fasepartikkel med et omgivende lag av motioner og et dobbelt elektrisk lag kalles micelle.
Regler for å skrive kolloidale miceller:


1-1 ladeelektrolytt
T – dispergert fasepartikkel.
AA er grensen mellom de tette og diffuse delene.
BB – glidende grense.
Skyvegrensen kan eller ikke falle sammen med linje AA.
pH-verdien der zetapotensialet er null kalles isoelektrisk punkt.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2 NaCl
1. Overskudd av CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n - x)Cl - ) 2 x + x Cl - - micellenotasjon.
CaSO 4 m – tilslag.
CaSO 4 m∙nCa 2+ – kjerne.
CaSO 4 m∙nCa 2+ 2( n - x)Cl - - partikkel.
2. Overskudd av Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micelle
CaSO 4 m – tilslag.
CaSO 4 m∙nSO 4 2 + – kjerne.
CaSO 4 m∙nSO 4 2- 2(n-x)Na+-partikkel
Gelemholtz-Smoluchowski-ligningen

 - lineær hastighet på grenseforskyvning (ved elektroosmose).
- lineær hastighet på grenseforskyvning (ved elektroosmose).
 - potensialforskjell over kondensatorplatene (ved elektroosmose).
- potensialforskjell over kondensatorplatene (ved elektroosmose).


 - volumetrisk strømningshastighet for løsningen, S– tverrsnittsareal av cellen.
- volumetrisk strømningshastighet for løsningen, S– tverrsnittsareal av cellen.

E– elektrisk feltstyrke.


 (for elektroosmose).
(for elektroosmose).
For strømningspotensial:

 - potensiell
- potensiell
 - trykk på membranen
- trykk på membranen
Som regel er verdiene for elektroforetiske mobiliteter og elektroosmotiske mobiliteter mindre enn de beregnede. Dette skjer på grunn av:
Avslappende effekt (når en dispergert fasepartikkel beveger seg, brytes symmetrien til den ioniske atmosfæren).
Elektroforetisk hemming (forekomsten av ytterligere friksjon som følge av bevegelsen av motioner).
Forvrengning av strømlinjer ved elektrisk ledende partikler.
Sammenheng mellom overflatespenning og potensial. Lippmann ligning.
Dannelsen av EDL skjer spontant på grunn av systemets ønske om å redusere overflateenergien. Under konstante forhold T Og s den generaliserte ligningen for termodynamikkens første og andre lov ser slik ut:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1. Lippmann-ligning.
- 1. Lippmann-ligning.
 - overflateladningstetthet.
- overflateladningstetthet.
 - differensiell kapasitans.
- differensiell kapasitans.
 - 2. Lippmann-ligning.
- 2. Lippmann-ligning.
MED– kapasitet.
La oss løse den første Lippmann-ligningen og den fundamentale adsorpsjonsligningen:
 ,
,


 , Deretter
, Deretter
 - Nernst-ligningen
- Nernst-ligningen
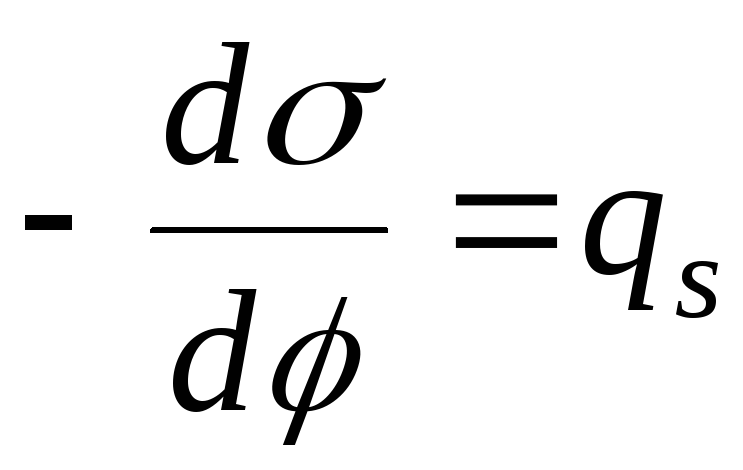 ,
,
 ,
,




 - ligning av den elektrokapillære kurven (ECC).
- ligning av den elektrokapillære kurven (ECC).
I  :
: , Men
, Men 
Kationiske overflateaktive stoffer (CPAS) reduserer den katodiske grenen til EKC.
Anioniske overflateaktive stoffer (APS) reduserer den anodiske grenen til EKC.
Ikke-ioniske overflateaktive stoffer (NSAS) reduserer den midtre delen av ECC.
Stabilitet av spredte systemer. Usammenhengende press.
Dispergerte systemer kan deles inn:
Systemer som er termodynamisk ustabile kan være kinetisk stabile på grunn av overgangen til en metastabil tilstand.
Det er to typer stabilitet:
Sedimentasjonsstabilitet (i forhold til tyngdekraften).
Aggregativ stabilitet. (i forhold til vedheft)
Koagulasjon er en prosess med partikkeladhesjon, som fører til tap av aggregatstabilitet. Koagulasjon kan være forårsaket av temperaturendringer, pH, omrøring og ultralyd.
Koagulasjon skiller seg ut:
Vendbar.
Irreversibel.
Koagulasjon oppstår ved innføring av elektrolytter.
Koagulasjonsregler:

Film- dette er den delen av systemet som ligger mellom to grensesnittflater.
Usammenhengende press oppstår når filmtykkelsen avtar kraftig som følge av samspillet mellom overflatelag som nærmer seg.

"-" - når filmtykkelsen avtar, øker det usammenhengende trykket.
P 0 er trykket i bulkfasen, som er en fortsettelse av mellomsjiktet.
P 1 – trykk i filmen.
Teori om stabilitet. DLFO (Deryagin, Landau, Fairway, Overbeck).
I følge DLFO-teorien har usammenhengende trykk to komponenter:
Elektrostatisk PE (positiv, det er på grunn av kreftene til elektrostatisk frastøtning). Tilsvarer en reduksjon i Gibbs-energien med økende filmtykkelse.
Molekylær P M (negativ, på grunn av virkningen av attraktive krefter). Det er forårsaket av filmkomprimering på grunn av kjemiske overflatekrefter, virkningsradiusen til krefter er tiendedeler av nm med en energi på ca. 400 kJ/mol.
Total interaksjonsenergi:

 - systemet er samlet sett stabilt
- systemet er samlet sett stabilt
 - ustabilt system
- ustabilt system
P  positiv komponent.
positiv komponent.
Økningen skyldes en økning i potensiell energi når tynne filmer komprimeres. For filmer med stor tykkelse kompenseres overskuddet av ioneenergi og er lik energiinteraksjonen i volumet av dispersjonsmediet.
Hvis  (
( - filmtykkelse,
- filmtykkelse,  - ioneradius) tynning av filmen fører til forsvinning og reduksjon av molekyler og ioner med minimal overflateenergi i den. Antallet nabopartikler avtar, som et resultat av at den potensielle energien til partiklene som er igjen i filmen øker.
- ioneradius) tynning av filmen fører til forsvinning og reduksjon av molekyler og ioner med minimal overflateenergi i den. Antallet nabopartikler avtar, som et resultat av at den potensielle energien til partiklene som er igjen i filmen øker.


DLVO-teorien anser interaksjonen mellom partikler som interaksjonen mellom plater.

Partikler samhandler ikke



 - Laplace-ligningen,
- Laplace-ligningen,  ,
,


For svakt ladede overflater
For svært ladede overflater:

Den molekylære komponenten er interaksjonen mellom to atomer:
 ~
~
Interaksjon mellom et atom og en overflate:

La oss ta to poster:
D  For å oppnå den molekylære komponenten, er det nødvendig å summere alle interaksjonsenergiene til atomene på høyre og venstre plate.
For å oppnå den molekylære komponenten, er det nødvendig å summere alle interaksjonsenergiene til atomene på høyre og venstre plate.
Hvor  - Hamaker-konstant (tar hensyn til naturen til samvirkende kropper).
- Hamaker-konstant (tar hensyn til naturen til samvirkende kropper).
At. interaksjonsenergien til partikler i et system kan uttrykkes ved hjelp av potensielle kurver.
I – primærpotensial minimum. Dette er en sone med irreversibel koagulasjon, tiltrekningskreftene råder.
II - sone med aggregert stabilitet, frastøtende krefter dominerer.
III – sekundært potensial minimum (eller flokkuleringssone). Det er et elektrolyttlag mellom partiklene i den dispergerte fasen, og partiklene kan separeres og overføres til sonen for aggregeringsstabilitet.

Kurve 1 – systemet er samlet sett stabilt.
Kurve 2 – stabil i sone I, ustabil i sone II.
Kurve 3 – koagulasjon har oppstått i systemet.
Kurve 4 – ved punkt 4 den totale interaksjonsenergien U=0,  , tilsvarer dette ekstremumpunktet begynnelsen av rask koagulasjon.
, tilsvarer dette ekstremumpunktet begynnelsen av rask koagulasjon.
Det er to tilfeller:
1. Lett ladede overflater:
U = U E + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - dette er tykkelsen på laget som tilsvarer begynnelsen av koagulasjonsprosessen.
- dette er tykkelsen på laget som tilsvarer begynnelsen av koagulasjonsprosessen.






 - for svakt ladede overflater
- for svakt ladede overflater
 Deretter
Deretter 
2. For svært ladede overflater:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
La oss kvadre (3)

Koagulasjon:
Ved spesifikk adsorpsjon kan ioner adsorberes i superekvivalente mengder slik at overflaten kan endre ladningen. Overflaten lades opp igjen.
Ved spesifikk adsorpsjon kan ikke bare ioner med motsatte fortegn adsorberes, men også av samme fortegn.
Hvis ioner med samme tegn som overflaten adsorberes, vil det ikke være en dråpe i potensialet i overflatelaget, men en økning i det.

Nøytraliseringskoagulering (oppstår med deltakelse av svakt ladede partikler og avhenger ikke bare av ladningen til elektrolytt-koagulatoren, men også av potensialet ved grensen til de tette og diffuse lagene).
Smoluchowskis teori om rask koagulasjon.

Avhengighet av koagulasjonshastighet på elektrolyttkonsentrasjon.
I – koagulasjonshastigheten er lav,
II – koagulasjonshastigheten er nesten proporsjonal med elektrolyttkonsentrasjonen.
III – område med rask koagulasjon, hastigheten er praktisk talt uavhengig av konsentrasjon.
Grunnleggende bestemmelser:
Den opprinnelige solen er monodispers, lignende partikler har en sfærisk form.
Alle partikkelkollisjoner er effektive.
Når to primærpartikler kolliderer, dannes det en sekundærpartikkel. Sekundær + primær = tertiær. Primær, sekundær, tertiær – multiplisitet.
Når det gjelder kjemisk kinetikk, kan koagulasjonsprosessen beskrives med ligningen:

Løsningen vil være ligningen: 
 - halv koagulasjonstid. Dette er tiden hvor antall solpartikler reduseres med 2 ganger.
- halv koagulasjonstid. Dette er tiden hvor antall solpartikler reduseres med 2 ganger.

 ,
,
 ,
,
 ,
,


Når multiplisiteten øker, skifter maksimum av koagulasjonskurvene mot større verdier  .
.
Feil:
Antagelse om monodispersitet.
Antakelse om effektiviteten av alle kollisjoner.