4.5.b. Oksidativ spaltning av alkener
Under oksidasjon av alkener med en alkalisk vandig løsning av kaliumpermanganat ved oppvarming eller med en løsning av KMnO 4 i vandig svovelsyre, samt under oksidasjon av alkener med en løsning av krom (VI) oksid CrO 3 i eddiksyre eller kaliumdikromat og svovelsyre, gjennomgår den opprinnelig dannede glykolen oksidativ ødeleggelse. Sluttresultatet er spaltning av karbonskjelettet på stedet for dobbeltbindingen og dannelse av ketoner og/eller karboksylsyrer som sluttprodukter, avhengig av substituentene på dobbeltbindingen. Hvis begge karbonatomene ved dobbeltbindingen inneholder bare en alkylgruppe, vil sluttproduktet av uttømmende oksidasjon være en blanding av karboksylsyrer; alkenet tetrasubstituert ved dobbeltbindingen oksideres til to ketoner. Monosubstituerte alkener med en terminal dobbeltbinding spaltes til karboksylsyre og karbondioksid.

På grunn av de lave utbyttene av karboksylsyrer og ketoner, har reaksjonene av uttømmende oksidasjon av alkener i den klassiske versjonen ikke funnet bred anvendelse og ble tidligere hovedsakelig brukt til å bestemme strukturen til startalkenet fra produktene av destruktiv oksidasjon. For tiden utføres oksidasjonen av alkener (R-CH=CH-R og R-CH=CH 2) til karboksylsyrer (RCOOH) ved bruk av kaliumpermanganat eller dikromat under faseoverføringskatalyse. Utbyttet av karboksylsyrer overstiger 90%.

4.5.v. Ozonolyse av alkener
Reaksjonen av alkener med ozon er den viktigste metoden for oksidativ spaltning av alkener ved dobbeltbindingen. I mange tiår fungerte denne reaksjonen som hovedmetoden for å bestemme strukturen til utgangshydrokarbonet, og fant også bruk i syntesen av forskjellige karbonylforbindelser. Reaksjonen av et alken med ozon utføres ved å føre en strøm på ~5 % blanding av ozon og oksygen inn i en løsning av alkenet i metylenklorid eller etylacetat ved -80 0 -100 0 C. Fullføringen av reaksjonen er kontrollert av en test for fritt ozon med kaliumjodid. Mekanismen for denne unike og komplekse reaksjonen er etablert hovedsakelig takket være arbeidet til R. Krige. Det første produktet av 1,3-dipolar sykloaddisjon til en dobbeltbinding er det såkalte molozonidet (1,2,3-trioksolan). Dette adduktet er ustabilt og spaltes videre spontant for å åpne ringen og danne det normale ozonid (1,2,4-trioxolane) som sluttprodukt.

Det er nå generelt akseptert at transformasjonen av molozonid til vanlig ozonid skjer gjennom spaltningsmekanismen - rekombinasjon. Molozonid gjennomgår spontan åpning av den ustabile 1,2,3-trioksolanringen for å danne en karbonylforbindelse og et bipolart ion, som deretter reagerer med hverandre også i henhold til det 1,3-dipolare sykloaddisjonsskjemaet.

Ovennevnte skjema for omorganisering av molozonid til et normalt ozonid bekreftes av det faktum at hvis, før den fullstendige dannelsen av ozonidet, en annen karbonylforbindelse er tilstede i reaksjonsblandingen som en "interceptor" av det bipolare ion, så -kalt "blandet ozonid" dannes. For eksempel med ozonylisering cis-stilben i nærvær av benzaldehyd merket med 18 O-isotopen, etiketten er en del av eteren i stedet for peroksidbroen til ozonidet:
Dette resultatet er i god overensstemmelse med dannelsen av et blandet ozonid ved rekombinasjon av et bipolart ion med merket benzaldehyd:

Ozonider er svært ustabile forbindelser som spaltes eksplosivt. De er ikke isolert individuelt, men brytes ned av handlingen til et bredt utvalg av regenter. Det er nødvendig å skille mellom reduktiv og oksidativ spaltning. Under hydrolyse brytes ozonider sakte ned til karbonylforbindelser og hydrogenperoksid. Hydrogenperoksid oksiderer aldehyder til karboksylsyrer. Dette er den såkalte oksidative nedbrytningen av ozonider:

Under den oksidative nedbrytningen av ozonider dannes det således karboksylsyrer og (eller) ketoner, avhengig av strukturen til det opprinnelige alkenet. Luftoksygen, hydrogenperoksid, persyrer eller sølvhydroksid kan brukes som oksidasjonsmidler. Oftest i syntetisk praksis brukes hydrogenperoksyd i eddik eller maursyre, samt hydrogenperoksyd i et alkalisk medium, til dette formålet.


I praksis brukes metoden for oksidativ dekomponering av ozonider hovedsakelig for å oppnå karboksylsyrer.
Den reduktive spaltningen av ozonider er viktigere. De mest brukte reduksjonsmidlene er sink og eddiksyre, trifenylfosfin eller dimetylsulfid. I dette tilfellet er sluttproduktene av ozonolyse aldehyder eller ketoner, avhengig av strukturen til det opprinnelige alkenet.



Fra eksemplene ovenfor er det klart at en alken tetrasubstituert ved dobbeltbindingen under ozonolyse og påfølgende reduktiv dekomponering av ozonidet danner to ketoner, mens en trisubstituert alken gir et keton og et aldehyd. En disubstituert symmetrisk alken produserer to aldehyder under ozonolyse, og alkener med en terminal binding danner et aldehyd og formaldehyd.
En interessant modifikasjon av ozonolyse er en metode hvor natriumborhydrid brukes som ozonid-reduksjonsmiddel.I dette tilfellet er de endelige reaksjonsproduktene primære eller sekundære alkoholer dannet under reduksjonen av henholdsvis aldehyder og xtoner.


Ozonolyse av alkener er en kompleks, arbeidskrevende og eksplosiv prosess som krever bruk av spesialutstyr. Av denne grunn er det utviklet andre metoder for oksidativ spaltning av alkener til karbonylforbindelser og karboksylsyrer, som vellykket erstatter ozonolysereaksjonen i syntetisk praksis.
En av de moderne preparative metodene for oksidativ ødeleggelse av alkener ble foreslått i 1955 av R. Lemieux. Denne metoden er basert på hydroksylering av alkener med kaliumpermanganat, etterfulgt av spaltning av vicinal glykol med natriumperjodat NaIO 4 ved pH ~ 7 8. Periodatet i seg selv reagerer ikke med alkenet. Produktene av denne to-trinns oksidative spaltningen er ketoner eller karboksylsyrer, siden aldehyder også oksideres til karboksylsyrer under disse forholdene. I Lemieuxs metode oppstår ikke det tidkrevende problemet med å separere et av reaksjonsproduktene, mangandioksid, siden både dioksidet og manganatet igjen oksideres av perjodat til permanganationet. Dette tillater bare å bruke katalytiske mengder kaliumpermanganat. Nedenfor er noen typiske eksempler på oksidativ spaltning av alkener ved bruk av Lemieux-metoden.

Citronellol, en alkohol som finnes i roseolje, geraniumolje og sitronolje, oksideres av en blanding av kaliumpermanganat og natriumperjodat i vandig aceton ved 5-10 0 C til 6-hydroksy-4-metylheksankarboksylsyre med et kvantitativt utbytte.

I en annen variant av denne metoden brukes katalytiske mengder osmiumtetroksid i stedet for kaliumpermanganat (Lemieux og Johnson 1956). En spesiell fordel med kombinasjonen av OsO 4 og NaIO 4 er at den lar deg stoppe oksidasjon på aldehydstadiet. Osmiumtetroksid legger til dobbeltbindingen til en alken for å danne osmat, som oksideres av natriumperjodat til karbonylforbindelser for å regenerere osmiumtetroksid.

I stedet for osmiumtetroksid kan også rutheniumtetroksid RuO 4 brukes. Oksidativ ødeleggelse av alkener ifølge Lemieux-Johnson fører til de samme produktene som ozonolyse med reduktiv spaltning av ozonider.

Når det gjelder moderne organisk kjemi, betyr dette at kombinasjonen OsO 4 -NaIO 4 er syntetisk ekvivalent reaksjoner av ozonolyse av alkener etterfulgt av reduktiv spaltning. Likeledes er oksidasjonen av alkener med en blanding av permanganat og perjodat den syntetiske ekvivalenten til ozonolyse med oksidativ nedbrytning av ozonider.
Dermed er oksidasjon av alkener ikke bare et sett med forberedende metoder for fremstilling av alkoholer, epoksider, dioler, aldehyder, ketoner og karboksylsyrer, det er også en av de mulige måtene å bestemme strukturen til det opprinnelige alkenet. Således, i henhold til resultatet av den oksidative ødeleggelsen av et alken, kan posisjonen til dobbeltbindingen i molekylet bestemmes, mens det stereokjemiske resultatet syn- eller anti- Hydroksylering av en alken lar oss trekke konklusjoner om dens geometri.
4.5. Oksidasjon av alkener
Det er tilrådelig å dele oksidasjonsreaksjonene til alkener i to store grupper: reaksjoner der karbonskjelettet er bevart og reaksjoner med oksidativ ødeleggelse av karbonskjelettet til molekylet ved dobbeltbindingen. Den første gruppen av reaksjoner inkluderer epoksidering, så vel som hydroksylering, som fører til dannelse av vicinale dioler (glykoler). Når det gjelder sykliske alkener, produserer hydroksylering vicinal transe- eller cis-dioler. En annen gruppe inkluderer ozonolyse og uttømmende oksidasjonsreaksjoner av alkener, som fører til dannelse av ulike typer karbonylforbindelser og karboksylsyrer.
4.5.a. Oksidasjonsreaksjoner av alkener med bevaring av karbonskjelettet
1. Epoksidasjon (reaksjon av N.A. Prilezhaev, 1909)
Asykliske og sykliske alkener, når de reagerer med persyrer (persyrer) RCOOOH i et ikke-polart, likegyldig miljø, danner epoksider (oksiraner), derfor kalles selve reaksjonen epoksidasjonsreaksjonen.
I følge moderne nomenklatur IUPAC- en treleddet ring med ett oksygenatom kalles oksiran.
Epoksidering av alkener bør betraktes som en synkron, koordinert prosess der ioniske mellomprodukter som hydroksylkationen OH+ ikke deltar. Med andre ord er epoksidering av alkener en prosess syn-binding av ett oksygenatom til en dobbeltbinding med fullstendig bevaring av konfigurasjonen av substituenter ved dobbeltbindingen.
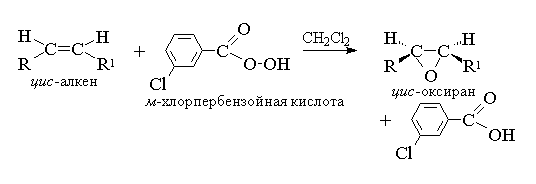

En mekanisme som er karakteristisk for samordnede prosesser har blitt foreslått for epoksidering.

Siden angrep av dobbeltbindingen av oksygenatomet til persyren er like sannsynlig på begge sider av dobbeltbindingsplanet, er de resulterende oksiranene enten meso-former eller blandinger av enantiomerer. Følgende persyrer brukes som epoksideringsmidler: perbenzoic, m-klorperbenzosyre, monoperftalsyre, pereddiksyre, trifluorpereddiksyre og performisk. Persyrer av den aromatiske serien brukes i form av individuelle reagenser, mens persyrer av den alifatiske serien - CH 3 CO 3 H, CF 3 CO 3 H og HCO 3 H ikke isoleres i individuell form, men brukes etter dannelse i interaksjonen mellom 30% eller 90% hydrogenperoksid og den tilsvarende karboksylsyren. Perbenzoin og m-klorperbenzosyre oppnås ved oksidasjon av benzosyre og m-klorbenzosyrer med 70% hydrogenperoksyd i en løsning av metansulfonsyre eller fra syrekloridene av disse syrene og hydrogenperoksyd.


Monoperftalsyre oppnås ved en lignende metode fra ftalsyreanhydrid og 30 % hydrogenperoksid.

Opprinnelig ble perbenzosyre eller monoperftalsyre brukt for å oppnå oksiraner (epoksider):

For tiden er den mest brukte for epoksidering m-klorperbenzosyre. I motsetning til andre persyrer er den holdbar i lang tid (opptil 1 år) og er helt sikker å håndtere. Utbytter av oksiraner oppnådd ved oksidasjon av asykliske og sykliske alkener m-klorperbenzosyre i en løsning av metylenklorid, kloroform eller dioksan er vanligvis ganske høye.



Persyrer genereres ofte direkte i en reaksjonsblanding av 90 % hydrogenperoksid og karboksylsyre i metylenklorid.


Alkener med en dobbeltbinding konjugert til en karbonylgruppe eller annen akseptorsubstituent er inaktive og for deres oksidasjon er det bedre å bruke sterkere oksidasjonsmidler, slik som trifluorpereddiksyre, oppnådd fra trifluoreddiksyreanhydrid og 90 % hydrogenperoksid i metylenklorid. Det enkleste oksiranet, etylenoksid, produseres industrielt ved oksidasjon av etylen med oksygen i nærvær av sølv som katalysator.

2. anti- Hydroksylering
Den tre-leddede ringen av oksiraner åpnes lett under påvirkning av en rekke nukleofile reagenser. Disse reaksjonene vil bli diskutert i detalj i avsnittet om asykliske og sykliske etere. Her vil bare hydrolyse av oksiraner bli vurdert. Hydrolysen av oksiraner katalyseres av både syrer og baser. I begge tilfeller dannes vicinale dioler, dvs. glykoler. I syrekatalyse, i det første trinnet, protoneres oksyran-oksygenatomet for å danne et syklisk oksoniumkation, som åpner seg som et resultat av det nukleofile angrepet av et vannmolekyl:

Nøkkeltrinnet i ringåpning, som bestemmer hastigheten på hele prosessen, er det nukleofile angrepet av vann på den protonerte formen av oksiran. Mekanistisk ligner denne prosessen åpningen av et bromion ved nukleofilt angrep av et bromidion eller et annet nukleofilt middel. Fra disse posisjonene bør det stereokjemiske resultatet være formasjonen transe-glykoler under nedbrytningen av sykliske epoksider. Faktisk, under den syrekatalyserte hydrolysen av cykloheksenoksid eller cyklopentenoksid, utelukkende transe-1,2-dioler.

Dermed tilsvarer to-trinns prosessen med alkenepoksidasjon etterfulgt av sur hydrolyse av epoksidet totalt reaksjonen anti-hydroksylering av alkener.
Begge stadier anti-Hydroksylering av alkener kan kombineres dersom alkenet behandles med vandig 30-70 % hydrogenperoksid i maur- eller trifluoreddiksyre. Begge disse syrene er sterke nok til å forårsake åpning av oksiranringen.

Basekatalysert åpning av oksiranringen fører også til dannelse av syklisk transe-glykoler.

Derfor er to-trinns prosessen med epoksidering av alkener etterfulgt av alkalisk hydrolyse av epoksider også en reaksjon anti-hydroksylering av alkener.
3. syn- Hydroksylering
Noen salter og oksider av overgangsmetaller i høyere oksidasjonstilstander er effektive reagenser syn-hydroksylering av dobbeltbindingen til en alken, når begge hydroksylgruppene legges til samme side av dobbeltbindingen. Oksidasjon av alkener med kaliumpermanganat er en av de eldste metodene syn Hydroksylering av en dobbeltbinding fortsetter å være mye brukt til tross for dens iboende begrensninger. Cis-1,2-cykloheksandiol ble først oppnådd av V.V. Markovnikov i 1878 ved hydroksylering av cykloheksen med en vandig løsning av kaliumpermanganat ved 0 0 C.

Denne metoden ble senere utviklet i verkene til den russiske forskeren E.E. Wagner altså syn-Hydroksylering av alkener under påvirkning av en vandig løsning av kaliumpermanganat kalles Wagner-reaksjonen. Kaliumpermanganat er et sterkt oksidasjonsmiddel som ikke bare kan hydroksylere dobbeltbindingen, men også spalte den resulterende vicinale diolen. For å unngå ytterligere nedbrytning av glykoler så mye som mulig, må reaksjonsbetingelsene kontrolleres nøye. Utbyttet av glykoler er vanligvis lave (30-60%). De beste resultatene oppnås ved hydroksylering av alkener i et lett alkalisk miljø (pH ~ 8 9) ved 0-5 0 C med en fortynnet 1 % vandig løsning av KMnO 4.


Til å begynne med produserer oksidasjonen av alkener med kaliumpermanganat en syklisk ester av mangansyre, som umiddelbart hydrolyseres til en vicinal diol.

Den sykliske esteren av mangansyre som et mellomprodukt ble ikke isolert, men dannelsen følger av eksperimenter med merket 18 O kaliumpermanganat: begge oksygenatomene i glykol viser seg å være merket under oksidasjonen av alkenet KMn 18 O 4. Dette betyr at begge oksygenatomene overføres fra oksidasjonsmidlet, og ikke fra løsningsmidlet - vann, som er i god overensstemmelse med den foreslåtte mekanismen.
En annen metode syn-hydroksylering av alkener under påvirkning av osmium (VIII) oksid OsO 4 ble foreslått av R. Kriege i 1936. Osmiumtetroksid er et fargeløst, flyktig, krystallinsk stoff, svært løselig i eter, dioksan, pyridin og andre organiske løsningsmidler. Når osmiumtetroksid reagerer med alkener i eter eller dioksan, dannes et svart bunnfall av syklisk osmisk syreester - osmat, som lett kan isoleres i individuell form. Tilsetningen av OsO 4 til dobbeltbindingen akselereres merkbart i oppløsning i pyridin. Dekomponeringen av osmater til vicinale glykoler oppnås ved virkningen av en vandig løsning av natriumhydrosulfitt eller hydrogensulfid.

Produktutbytte syn-Hydroksylering av alkener i denne metoden er betydelig høyere enn ved bruk av permanganat som oksidasjonsmiddel. En viktig fordel med Krige-metoden er fraværet av produkter av oksidativ spaltning av alkener, som er karakteristisk for permanganatoksidasjon.


Osmiumtetroksid er et veldig dyrt og vanskelig å få tak i, og det er også giftig. Derfor brukes osmium (VIII) oksid i syntesen av små mengder vanskelig tilgjengelige stoffer for å oppnå det høyeste utbyttet av diol. For å forenkle syn-hydroksylering av alkener under påvirkning av OsO 4, ble det utviklet en teknikk som tillater bruk av kun katalytiske mengder av dette reagenset. Hydroksylering av alkener utføres ved bruk av hydrogenperoksid i nærvær av OsO 4, for eksempel:

For å avslutte denne delen presenterer vi de stereokjemiske forholdene mellom alkenet cis- eller transe-konfigurasjon og konfigurasjonen av den resulterende vicinale diolen, som kan være cis- eller transe-isomer, erythro- eller trio-form, meso- eller D,L-form avhengig av substituentene i alkenet:

Lignende stereokjemiske forhold observeres i andre reaksjoner syn- eller anti- tilsetning ved flere bindinger av hydrogen, hydrogenhalogenider, vann, halogener, borhydrider og andre reagenser.
18. Redoksreaksjoner (forts. 2)
18.9. OVR som involverer organiske stoffer
I ORR av organiske stoffer med uorganiske stoffer er organiske stoffer oftest reduksjonsmidler. Når organisk materiale brenner i overflødig oksygen, dannes det alltid karbondioksid og vann. Reaksjoner er mer kompliserte ved bruk av mindre aktive oksidasjonsmidler. Denne delen diskuterer bare reaksjonene til representanter for de viktigste klassene av organiske stoffer med noen uorganiske oksidasjonsmidler.
Alkenes. Ved mild oksidasjon omdannes alkener til glykoler (toverdige alkoholer). De reduserende atomene i disse reaksjonene er karbonatomer forbundet med en dobbeltbinding.
Reaksjonen med en løsning av kaliumpermanganat skjer i et nøytralt eller svakt alkalisk medium som følger:
C 2 H 4 + 2KMnO 4 + 2H 2 O CH 2 OH–CH 2 OH + 2MnO 2 + 2KOH (kjøling)
Under mer alvorlige forhold fører oksidasjon til brudd på karbonkjeden ved dobbeltbindingen og dannelse av to syrer (i et sterkt alkalisk miljø - to salter) eller en syre og karbondioksid (i et sterkt alkalisk miljø - et salt og et karbonat):
1) 5CH 3 CH=CHCH 2 CH 3 + 8KMnO 4 + 12H 2 SO 4 5CH 3 COOH + 5C 2 H 5 COOH + 8MnSO 4 + 4K 2 SO 4 + 17H 2 O (oppvarming)
2) 5CH 3 CH=CH 2 + 10KMnO 4 + 15H 2 SO 4 5CH 3 COOH + 5CO 2 + 10MnSO 4 + 5K 2 SO 4 + 20H 2 O (oppvarming)
3) CH 3 CH=CHCH 2 CH 3 + 6KMnO 4 + 10KOH CH 3 COOK + C 2 H 5 COOK + 6H 2 O + 6K 2 MnO 4 (oppvarming)
4) CH 3 CH=CH 2 + 10KMnO 4 + 13KOH CH 3 COOK + K 2 CO 3 + 8H 2 O + 10K 2 MnO 4 (oppvarming)
Kaliumdikromat i et svovelsyremedium oksiderer alkener på samme måte som reaksjon 1 og 2.
Alkyner. Alkyner begynner å oksidere under litt mer alvorlige forhold enn alkener, så de oksiderer vanligvis ved å bryte karbonkjeden ved trippelbindingen. Som i tilfellet med alkaner, er de reduserende atomene her karbonatomer, i dette tilfellet forbundet med en trippelbinding. Som et resultat av reaksjonene dannes syrer og karbondioksid. Oksidasjon kan utføres med kaliumpermanganat eller dikromat i et surt miljø, for eksempel:
5CH 3 C CH + 8KMnO 4 + 12H 2 SO 4 5CH 3 COOH + 5CO 2 + 8MnSO 4 + 4K 2 SO 4 + 12H 2 O (oppvarming)
Noen ganger er det mulig å isolere mellomliggende oksidasjonsprodukter. Avhengig av plasseringen av trippelbindingen i molekylet er disse enten diketoner (R 1 –CO–CO–R 2) eller aldoketoner (R–CO–CHO).
Acetylen kan oksideres med kaliumpermanganat i et lett alkalisk medium til kaliumoksalat:
3C 2 H 2 + 8KMnO 4 = 3K 2 C 2 O 4 + 2H 2 O + 8MnO 2 + 2KOH
I et surt miljø fortsetter oksidasjonen til karbondioksid:
C 2 H 2 + 2KMnO 4 + 3H 2 SO 4 = 2CO 2 + 2MnSO 4 + 4H 2 O + K 2 SO 4
Benzenhomologer. Benzenhomologer kan oksideres med en løsning av kaliumpermanganat i et nøytralt miljø til kaliumbenzoat:
C 6 H 5 CH 3 + 2KMnO 4 = C 6 H 5 COOK + 2MnO 2 + KOH + H 2 O (ved koking)
C 6 H 5 CH 2 CH 3 + 4KMnO 4 = C 6 H 5 COOK + K 2 CO 3 + 2H 2 O + 4MnO 2 + KOH (ved oppvarming)
Oksidasjon av disse stoffene med kaliumdikromat eller permanganat i et surt miljø fører til dannelse av benzosyre.
Alkoholer. Det direkte oksidasjonsproduktet av primære alkoholer er aldehyder, og oksidasjonsproduktene til sekundære alkoholer er ketoner.
Aldehyder dannet under oksidasjon av alkoholer oksideres lett til syrer, derfor oppnås aldehyder fra primære alkoholer ved oksidasjon med kaliumdikromat i et surt medium ved aldehydets kokepunkt. Når aldehyder fordamper, har de ikke tid til å oksidere.
3C 2 H 5 OH + K 2 Cr 2 O 7 + 4H 2 SO 4 = 3CH 3 CHO + K 2 SO 4 + Cr 2 (SO 4) 3 + 7H 2 O (oppvarming)
Med et overskudd av oksidasjonsmiddel (KMnO 4, K 2 Cr 2 O 7) i ethvert miljø, oksideres primære alkoholer til karboksylsyrer eller deres salter, og sekundære alkoholer oksideres til ketoner. Tertiære alkoholer oksiderer ikke under disse forholdene, men metylalkohol oksideres til karbondioksid. Alle reaksjoner oppstår ved oppvarming.
Toverdig alkohol, etylenglykol HOCH 2 –CH 2 OH, ved oppvarming i et surt miljø med en løsning av KMnO 4 eller K 2 Cr 2 O 7, oksideres lett til karbondioksid og vann, men noen ganger er det mulig å isolere mellomprodukter (HOCH 2 –COOH, HOOC– COOH, etc.).
Aldehyder. Aldehyder er ganske sterke reduksjonsmidler, og oksideres derfor lett av forskjellige oksidasjonsmidler, for eksempel: KMnO 4, K 2 Cr 2 O 7, OH. Alle reaksjoner oppstår ved oppvarming:
3CH 3 CHO + 2KMnO 4 = CH 3 COOH + 2CH 3 COOK + 2MnO 2 + H 2 O
3CH 3 CHO + K 2 Cr 2 O 7 + 4H 2 SO 4 = 3CH 3 COOH + Cr 2 (SO 4) 3 + 7H 2 O
CH 3 CHO + 2OH = CH 3 COONH 4 + 2Ag + H 2 O + 3NH 3
Formaldehyd med et overskudd av oksidasjonsmiddel oksideres til karbondioksid.
18.10. Sammenligning av redoksaktiviteten til ulike stoffer
Fra definisjonene av begrepene "oksiderende atom" og "reduserende atom" følger det at atomer i høyeste oksidasjonstilstand kun har oksiderende egenskaper. Tvert imot har atomer i den laveste oksidasjonstilstanden kun reduserende egenskaper. Atomer i mellomliggende oksidasjonstilstander kan være både oksidasjonsmidler og reduksjonsmidler.
Samtidig, bare basert på graden av oksidasjon, er det umulig å entydig vurdere redoksegenskapene til stoffer. Som et eksempel, vurder forbindelsene til elementer i VA-gruppen. Nitrogen(V)- og antimon(V)-forbindelser er mer eller mindre sterke oksidasjonsmidler, vismut(V)-forbindelser er svært sterke oksidasjonsmidler, og fosfor(V)-forbindelser har praktisk talt ingen oksiderende egenskaper. I dette og andre lignende tilfeller er det som betyr noe hvor karakteristisk en gitt oksidasjonstilstand er for et gitt grunnstoff, det vil si hvor stabile er forbindelsene som inneholder atomer til et gitt grunnstoff i denne oksidasjonstilstanden.
Enhver redoksreaksjon fortsetter i retning av dannelsen av et svakere oksidasjonsmiddel og et svakere reduksjonsmiddel. I det generelle tilfellet kan muligheten for at enhver ORR oppstår, som enhver annen reaksjon, bestemmes av tegnet på endringen i Gibbs-energien. I tillegg, for å kvantifisere redoksaktiviteten til stoffer, brukes de elektrokjemiske egenskapene til oksidasjonsmidler og reduksjonsmidler (standardpotensialer for redokspar). Basert på disse kvantitative egenskapene er det mulig å konstruere serier med redoksaktivitet av forskjellige stoffer. Serien av metallspenninger som du kjenner er konstruert på akkurat denne måten. Denne serien gjør det mulig å sammenligne de reduserende egenskapene til metaller i vandige løsninger under standardforhold ( Med= 1 mol/l, T= 298,15 K), så vel som de oksiderende egenskapene til enkle akvakasjoner. Hvis du plasserer ioner (oksidasjonsmidler) i den øverste raden i denne raden, og metallatomer (reduksjonsmidler) i den nederste raden, vil venstre side av denne raden (før hydrogen) se slik ut:
I denne serien øker de oksiderende egenskapene til ioner (øverste linje) fra venstre til høyre, og de reduserende egenskapene til metaller (bunnlinje), tvert imot, fra høyre til venstre.
Tar man hensyn til forskjellene i redoksaktivitet i ulike miljøer, er det mulig å konstruere lignende serier for oksidasjonsmidler. Således, for reaksjoner i et surt miljø (pH = 0), oppnås en "fortsettelse" av serien av metallaktiviteter i retning av økende oksidative egenskaper
Som i metallaktivitetsserien øker i denne serien oksydasjonsegenskapene til oksidasjonsmidler (øverste linje) fra venstre til høyre. Men ved å bruke denne serien er det mulig å sammenligne reduksjonsaktiviteten til reduksjonsmidler (bunnlinjen) bare hvis deres oksiderte form faller sammen med den som er vist i den øverste linjen; i dette tilfellet forsterkes den fra høyre til venstre.
La oss se på noen få eksempler. For å finne ut om denne ORR er mulig, vil vi bruke en generell regel som bestemmer retningen for redoksreaksjoner (reaksjoner går i retning av dannelse av et svakere oksidasjonsmiddel og et svakere reduksjonsmiddel).
1. Er det mulig å redusere kobolt fra en CoSO 4 løsning med magnesium?
Magnesium er et sterkere reduksjonsmiddel enn kobolt, og Co 2 -ioner er sterkere oksidasjonsmidler enn Mg 2 -ioner, derfor er det mulig.
2. Er det mulig å oksidere kobber til CuCl 2 i et surt miljø med en løsning av FeCl 3?
Siden Fe 3B-ioner er sterkere oksidasjonsmidler enn Cu 2-ioner, og kobber er et sterkere reduksjonsmiddel enn Fe 2-ioner, er det mulig.
3. Er det mulig å få en løsning av FeCl 3 ved å blåse oksygen gjennom en løsning av FeCl 2 surgjort med saltsyre?
Det ser ikke ut til, siden oksygen i vår serie er til venstre for Fe 3 -ionene og er et svakere oksidasjonsmiddel enn disse ionene. Men i en vandig løsning reduseres oksygen nesten aldri til H 2 O 2, i dette tilfellet reduseres det til H 2 O og foregår mellom Br 2 og MnO 2. Derfor er en slik reaksjon mulig, selv om den går ganske sakte (hvorfor?).
4. Er det mulig å oksidere H 2 O 2 i et surt miljø med kaliumpermanganat?
I dette tilfellet er H 2 O 2 et reduksjonsmiddel og et sterkere reduksjonsmiddel enn Mn 2B-ioner, og MnO 4-ioner er sterkere oksidasjonsmidler enn oksygenet som dannes fra peroksid. Derfor er det mulig.
En lignende serie konstruert for ORR i et alkalisk medium er som følger:
I motsetning til "syre"-serien, kan ikke denne serien brukes sammen med metallaktivitetsserien.
Elektron-ion balansemetode (halvreaksjonsmetode), intermolekylær ORR, intramolekylær ORR, dismutasjon ORR (disproporsjonering, selvoksidasjon-selvreduksjon), ORR kommutering, passivering.
- Ved hjelp av elektron-ionbalansemetoden, komponer ligninger for reaksjonene som oppstår når a) H 2 S (S, mer presist, S 8 ) løsning tilsettes til en løsning av kaliumpermanganat surgjort med svovelsyre; b) KHS; c) K2S; d) H2SO3; e) KHSO 3; e) K2SO3; e) HNO2; g) KNO 2; i) KI (12); j) FeS04; l) C2H5OH (CH3COOH); m) CH3CHO; n) (COOH)2(C02); n) K2C204. Her og nedenfor, der det er nødvendig, er oksidasjonsprodukter angitt i krøllete parenteser.
- Skriv ned ligninger for reaksjonene som oppstår når følgende gasser føres gjennom en løsning av kaliumpermanganat surgjort med svovelsyre: a) C 2 H 2 (CO 2 ); b) C2H4 (CO2); c) C3H4 (propyn) (CO2 og CH3COOH); d) C3H6; e) CH4; e) HCHO.
- Det samme, men en reduksjonsmiddelløsning tilsettes til en nøytral løsning av kaliumpermanganat: a) KHS; b) K2S; c) KHSO 3; d) K2SO3; e) KNO 2; e) KI.
- Det samme, men en løsning av kaliumhydroksid er tidligere tilsatt til kaliumpermanganatløsningen: a) K 2 S (K 2 SO 4 ); b) K2SO3; c) KNO 2; d) KI (KIO 3).
- Skriv ned ligninger for følgende reaksjoner som oppstår i løsning: a) KMnO 4 + H 2 S ...;
b) KMn04 + HCl ...;
c) KMn04 + HBr ...;
d) KMnO 4 + HI ... - Lag følgende ligninger for ORR for mangandioksid:
- Løsninger av følgende stoffer ble tilsatt til en løsning av kaliumdikromat surgjort med svovelsyre: a) KHS; b) K2S; c) HNO2; d) KNO 2; e) KI; f) FeS04; g) CH3CH2CHO; i) H2SO3; j) KHSO 3; k) K 2 SO 3. Skriv ned ligningene for reaksjonene som finner sted.
- Det samme, men følgende gasser føres gjennom løsningen: a) H 2 S; b) SO 2.
- Løsninger av a) K2S (K2SO4); b) K2SO3; c) KNO 2; d) KI (KIO 3). Skriv ned ligningene for reaksjonene som finner sted.
- En løsning av kaliumhydroksid ble tilsatt til løsningen av krom(III)klorid inntil det opprinnelig dannede bunnfallet var oppløst, og deretter ble bromvann tilsatt. Skriv ned ligningene for reaksjonene som finner sted.
- Det samme, men på siste trinn ble tilsatt en løsning av kaliumperoksodisulfat K 2 S 2 O 8, som ble redusert til sulfat under reaksjonen.
- Skriv ned ligningene for reaksjonene som oppstår i løsningen:
- Skriv ned likninger for reaksjonene mellom fast kromtrioksid og følgende stoffer: a) C; b) CO; c) S (S02); d) H2S; e) NH3; e) C2H5OH (CO2 og H20); g) CH3COCH3.
- Skriv ned likninger for reaksjonene som oppstår når følgende stoffer tilsettes konsentrert salpetersyre: a) S (H 2 SO 4 ); b) P4((HPO3)4); c) grafitt; d) Se; e) I2 (HIO3); f) Ag; g) Cu; i) Pb; j) KF; l) FeO; m) FeS; m) MgO; n) MgS; p) Fe(OH)2; c) P203; t) As203 (H3AsO4); y) As 2S3; f) Fe(NO3)2; x) P4010; v) Cu2S.
- Det samme, men når de passerer følgende gasser: a) CO; b) H2S; c) N20; d) NH3; e) NEI; f) H2Se; g) HI.
- Reaksjonene vil forløpe likt eller annerledes i følgende tilfeller: a) et stykke magnesium ble plassert i et høyt reagensrør to tredjedeler fylt med konsentrert salpetersyre; b) en dråpe konsentrert salpetersyre ble plassert på overflaten av magnesiumplaten? Skriv ned reaksjonslikninger.
- Hva er forskjellen mellom reaksjonen av konsentrert salpetersyre med hydrogensulfidsyre og med gassformig hydrogensulfid? Skriv ned reaksjonslikninger.
- Vil ORR fortsette på samme måte når vannfritt krystallinsk natriumsulfid og dets 0,1 M løsning tilsettes til en konsentrert løsning av salpetersyre?
- En blanding av følgende stoffer ble behandlet med konsentrert salpetersyre: Cu, Fe, Zn, Si og Cr. Skriv ned ligningene for reaksjonene som finner sted.
- Skriv ned ligninger for reaksjonene som oppstår når følgende stoffer tilsettes fortynnet salpetersyre: a) I 2 ; b) Mg; c) Al; d) Fe; e) FeO; f) FeS; g) Fe(OH)2; i) Fe(OH)3; j) MnS; l) Cu2S; l) CuS; m) CuO; n) Na2Scr; p) Na2Sp; c) P4010.
- Hvilke prosesser vil skje når a) ammoniakk, b) hydrogensulfid, c) karbondioksid føres gjennom en fortynnet løsning av salpetersyre?
- Skriv ned ligninger for reaksjonene som oppstår når følgende stoffer tilsettes konsentrert svovelsyre: a) Ag; b) Cu; c) grafitt; d) HCOOH; e) C6H1206; f) NaCl cr; g) C2H5OH.
- Når hydrogensulfid føres gjennom kald konsentrert svovelsyre, dannes S og SO 2, varmkonsentrert H 2 SO 4 oksiderer svovel til SO 2. Skriv ned reaksjonslikninger. Hvordan vil reaksjonen forløpe mellom varmkonsentrert H 2 SO 4 og hydrogensulfid?
- Hvorfor oppnås hydrogenklorid ved å behandle krystallinsk natriumklorid med konsentrert svovelsyre, men hydrogenbromid og hydrogenjodid oppnås ikke ved denne metoden?
- Skriv ned ligninger for reaksjonene som oppstår under interaksjonen av fortynnet svovelsyre med a) Zn, b) Al, c) Fe, d) krom i fravær av oksygen, e) krom i luft.
- Skriv ned reaksjonsligninger som karakteriserer redoksegenskapene til hydrogenperoksid:
- Hvilke reaksjoner oppstår når følgende stoffer varmes opp: a) (NH 4) 2 CrO 4; b) NaN03; c) CaC03; d) Al(N03)3; e) Pb(NO3)3; f) AgN03; g) Hg(NO3)2; i) Cu(NO3)2; j) CuO; l) NaClO4; m) Ca(ClO4)2; m) Fe(NO3)2; n) PCl 5; p) MnCl4; c) H2C204; r) LiN03; y) HgO; f) Ca(NO3)2; x) Fe(OH)3; v) CuCl2; h) KC103; w) KC102; y) CrO3?
- Når varme løsninger av ammoniumklorid og kaliumnitrat kombineres, oppstår en reaksjon ledsaget av frigjøring av gass. Skriv en ligning for denne reaksjonen.
- Skriv ned ligninger for reaksjonene som oppstår når a) klor, b) bromdamp føres gjennom en kald løsning av natriumhydroksid. Det samme, men gjennom en varm løsning.
- Ved interaksjon med en varm konsentrert løsning av kaliumhydroksid, gjennomgår selen dismutering til nærmeste stabile oksidasjonstilstander (–II og +IV). Skriv en ligning for denne ORR.
- Under de samme forholdene gjennomgår svovel en lignende dismutasjon, men overskudd av svovel reagerer med sulfittioner for å danne tiosulfationer S 2 O 3 2. Skriv ned ligningene for reaksjonene som finner sted. ;
- Skriv ned ligninger for elektrolysereaksjonene til a) en løsning av kobbernitrat med en sølvanode, b) en løsning av blynitrat med en kobberanode.
a) CrCl2 + FeCl3; b) CrS04 + FeCl3; c) CrS04 + H2SO4 + O2;
d) CrS04 + H2SO4 + Mn02; e) CrSO 4 + H 2 SO 4 + KMnO 4.
I hvilken av disse reaksjonene er hydrogenperoksid et oksidasjonsmiddel, og i hvilket er det et reduksjonsmiddel?
| Erfaring 1. Oksidative egenskaper av kaliumpermanganat i et surt miljø. Til 3-4 dråper av en løsning av kaliumpermanganat, tilsett et like stort volum av en fortynnet løsning av svovelsyre, og deretter en løsning av natriumsulfitt til den er misfarget. Skriv en ligning for reaksjonen. Erfaring 2.Oksiderende egenskaper av kaliumpermanganat i et nøytralt miljø. Tilsett 5-6 dråper natriumsulfittløsning til 3-4 dråper kaliumpermanganatløsning. Hvilket stoff ble frigjort som et bunnfall? Erfaring 3. Oksidative egenskaper av kaliumpermanganat i et alkalisk miljø. Til 3-4 dråper kaliumpermanganatløsning tilsett 10 dråper konsentrert natriumhydroksidløsning og 2 dråper natriumsulfittløsning. Løsningen skal bli grønn. Erfaring 4. Oksidative egenskaper av kaliumdikromat i et surt miljø. Surgjør 6 dråper kaliumdikromatløsning med fire dråper fortynnet svovelsyreløsning og tilsett natriumsulfittløsning til fargen på blandingen endres. Erfaring 5. Oksiderende egenskaper for fortynnet svovelsyre. Plasser et sinkgranulat i det ene reagensglasset og et stykke kobbertape i det andre. Tilsett 8-10 dråper av en fortynnet svovelsyreløsning i begge reagensglassene. Sammenlign fenomenene som oppstår. GJENNOMFØR EKSPERIMENTET I EN DRUKKROK! Erfaring 6. Oksiderende egenskaper til konsentrert svovelsyre. Ligner på eksperiment 5, men tilsett en konsentrert løsning av svovelsyre. Et minutt etter starten av frigjøringen av gassformige reaksjonsprodukter, innfør strimler av filterpapir fuktet med løsninger av kaliumpermanganat og kobbersulfat i reagensrørene. Forklar fenomenene som oppstår. GJENNOMFØR EKSPERIMENTET I EN DRUKKROK! Erfaring 7. Oksiderende egenskaper for fortynnet salpetersyre. Ligner på eksperiment 5, men tilsett en fortynnet løsning av salpetersyre. Observer fargeendringen til de gassformige reaksjonsproduktene. GJENNOMFØR EKSPERIMENTET I EN DRUKKROK! Erfaring 8. Oksiderende egenskaper til konsentrert salpetersyre. Plasser et stykke kobbertape i et reagensrør og tilsett 10 dråper av en konsentrert løsning av salpetersyre. Varm forsiktig til metallet er helt oppløst. GJENNOMFØR EKSPERIMENTET I EN DRUKKROK! Erfaring 9. Oksiderende egenskaper av kaliumnitritt. Til 5-6 dråper av en løsning av kaliumnitritt, tilsett et like stort volum av en fortynnet løsning av svovelsyre og 5 dråper av en løsning av kaliumjodid. Hvilke stoffer dannes? Erfaring 10. Reduserende egenskaper av kaliumnitritt. Til 5-6 dråper av en løsning av kaliumpermanganat, tilsett et like stort volum av en fortynnet løsning av svovelsyre og en løsning av kaliumnitritt til blandingen er fullstendig misfarget. Erfaring 11.Termisk nedbrytning av kobbernitrat. Plasser en mikrospatel av kobbernitrattrihydrat i et reagensrør, fest den i et stativ og varm den forsiktig opp med åpen flamme. Observer dehydrering og påfølgende dekomponering av salt. GJENNOMFØR EKSPERIMENTET I EN DRUKKROK! Erfaring 12.Termisk nedbrytning av blynitrat. Utfør samme prosedyre som eksperiment 11, plasser blynitrat i et reagensrør. GJENNOMFØR EKSPERIMENTET I EN DRUKKROK! Hva er forskjellen mellom prosessene som skjer under nedbrytningen av disse saltene? |
Redoksreaksjoner som involverer organiske stoffer
Tendensen til organiske forbindelser til å oksidere er assosiert med tilstedeværelsen multiple bindinger, funksjonelle grupper, hydrogenatomer ved karbonatomet som inneholder den funksjonelle gruppen.
Den sekvensielle oksidasjonen av organiske stoffer kan representeres som følgende kjede av transformasjoner:
Mettet hydrokarbon → Umettet hydrokarbon → Alkohol → Aldehyd (keton) → Karboksylsyre → CO 2 + H 2 O
Det genetiske forholdet mellom klasser av organiske forbindelser er her representert som en rekke redoksreaksjoner som sikrer overgangen fra en klasse av organiske forbindelser til en annen. Det fullføres av produktene av fullstendig oksidasjon (forbrenning) av enhver representant for klassene av organiske forbindelser.
Avhengighet av redokskapasiteten til et organisk stoff på strukturen:
Den økte tendensen til organiske forbindelser til å oksidere skyldes tilstedeværelsen av stoffer i molekylet:
- flere bindinger(dette er grunnen til at alkener, alkyner og alkadiener så lett oksideres);
- visse funksjonsgrupper, som lett kan oksideres (–-SH, –OH (fenolisk og alkoholisk), – NH 2 ;
- aktiverte alkylgrupper, plassert ved siden av flere obligasjoner. For eksempel kan propen oksideres til det umettede aldehyd akrolein med atmosfærisk oksygen i nærvær av vanndamp på vismut-molybden-katalysatorer.
H 2 C═CH−CH 3 → H 2 C═CH−COH
Og også oksidasjon av toluen til benzosyre med kaliumpermanganat i et surt miljø.
5C 6 H 5 CH 3 + 6KMnO 4 + 9H 2 SO 4 → 5C 6 H 5 COOH + 3K 2 SO 4 + 6MnSO 4 +14H 2 O
- tilstedeværelsen av hydrogenatomer ved et karbonatom som inneholder en funksjonell gruppe.
Et eksempel er reaktiviteten i oksidasjonsreaksjonene til primære, sekundære og tertiære alkoholer ved oksidasjonsreaktivitet.
Til tross for at det under enhver redoksreaksjon oppstår både oksidasjon og reduksjon, klassifiseres reaksjoner avhengig av hva som skjer direkte med den organiske forbindelsen (hvis den er oksidert, snakker vi om oksidasjonsprosessen, hvis den reduseres, snakker vi om reduksjonsprosessen ).
Således, i reaksjonen av etylen med kaliumpermanganat, vil etylen oksideres, og kaliumpermanganat reduseres. Reaksjonen kalles etylenoksidasjon.
Bruken av konseptet "oksidasjonstilstand" (CO) i organisk kjemi er svært begrenset og implementeres først og fremst i utarbeidelse av ligninger for redoksreaksjoner. Men tatt i betraktning at en mer eller mindre konstant sammensetning av reaksjonsprodukter bare er mulig med fullstendig oksidasjon (forbrenning) av organiske stoffer, forsvinner det tilrådelig å arrangere koeffisienter i ufullstendige oksidasjonsreaksjoner. Av denne grunn er man vanligvis begrenset til å tegne et diagram over transformasjonene av organiske forbindelser.
Når vi studerte de komparative egenskapene til uorganiske og organiske forbindelser, ble vi kjent med bruken av oksidasjonstilstand (s.o.) (i organisk kjemi, først og fremst karbon) og metoder for å bestemme den:
1) beregning av gjennomsnitt s.o. karbon i et molekyl av organisk materiale:
-8/3 +1
Denne tilnærmingen er berettiget hvis under reaksjonen alle kjemiske bindinger i et organisk stoff blir ødelagt (forbrenning, fullstendig nedbrytning).
2) definisjon av s.o. hvert karbonatom:
I dette tilfellet er oksidasjonstilstanden til ethvert karbonatom i en organisk forbindelse lik den algebraiske summen av tallene for alle bindinger med atomer av mer elektronegative elementer, tatt i betraktning med "+"-tegnet på karbonatomet, og antall bindinger med hydrogenatomer (eller et annet mer elektropositivt element), tatt i betraktning med tegnet "-" ved karbonatomet. I dette tilfellet tas det ikke hensyn til bindinger med nabokarbonatomer.
Som et enkelt eksempel, la oss bestemme oksidasjonstilstanden til karbon i et metanolmolekyl.
![]()
Et karbonatom er koblet til tre hydrogenatomer (disse bindingene telles med et "–"-tegn), og en binding er koblet til et oksygenatom (det telles med et "+"-tegn). Vi får: -3 + 1 = -2. Dermed er oksidasjonstilstanden til karbon i metanol -2.
Den beregnede graden av oksidasjon av karbon, selv om den er en betinget verdi, indikerer arten av skiftet i elektrontetthet i molekylet, og endringen som et resultat av reaksjonen indikerer at redoksprosessen finner sted.
La oss avklare i hvilke tilfeller det er bedre å bruke en eller annen metode.
Prosessene med oksidasjon, forbrenning, halogenering, nitrering, dehydrogenering og dekomponering er klassifisert som redoksprosesser.
Når du flytter fra en klasse av organiske forbindelser til en annen Ogøke graden av forgrening av karbonskjelettet molekyler av forbindelser innenfor en egen klasse Oksydasjonstilstanden til karbonatomet som er ansvarlig for den reduserende evnen til forbindelsen endres.
Organiske stoffer hvis molekyler inneholder karbonatomer med maksimum(- og +) CO-verdier(-4, -3, +2, +3), inngå en fullstendig oksidasjons-forbrenningsreaksjon, men motstandsdyktig mot milde og middels oksiderende midler.
Stoffer hvis molekyler inneholder karbonatomer i CO-1; 0; +1, oksider lett, deres reduserende evner er nære, derfor kan deres ufullstendige oksidasjon oppnås på grunn av en av de kjente oksidasjonsmidler med lav og middels styrke. Disse stoffene kan vises dobbel natur, fungerer som et oksidasjonsmiddel, akkurat som det er iboende i uorganiske stoffer.
Når du skriver ligninger for reaksjonene ved forbrenning og nedbrytning av organiske stoffer, er det bedre å bruke gjennomsnittsverdien av d.o. karbon.
For eksempel:
![]()
La oss lage en komplett ligning for en kjemisk reaksjon ved å bruke balansemetoden.
Gjennomsnittlig verdi av karbonoksidasjonstilstand i n-butan:
Oksydasjonstilstanden til karbon i karbonmonoksid (IV) er +4.
La oss lage et elektronisk balansediagram:

Vær oppmerksom på den første halvdelen av elektronbalansen: karbonatomet har en brøkdel d.o. nevneren er 4, så vi beregner overføringen av elektroner ved å bruke denne koeffisienten.
De. overgangen fra -2,5 til +4 tilsvarer overgangen 2,5 + 4 = 6,5 enheter. Fordi 4 karbonatomer er involvert, da vil 6,5 · 4 = 26 elektroner totalt gis fra butankarbonatomene.
Med tanke på de funnet koeffisientene, vil ligningen for den kjemiske reaksjonen ved n-butanforbrenning se slik ut:
Du kan bruke metoden for å bestemme den totale ladningen av karbonatomer i et molekyl:
(4 C) -10 …… → (1 C) +4 , og tar i betraktning at antall atomer før og etter =-tegnet skal være det samme, utligner vi (4C) -10 …… →[(1 C) +4 ] · 4
Derfor innebærer overgangen fra -10 til +16 tap av 26 elektroner.
I andre tilfeller bestemmer vi verdiene til s.o. hvert karbonatom i forbindelsen, og ta hensyn til rekkefølgen for erstatning av hydrogenatomer ved primære, sekundære, tertiære karbonatomer:


Først skjer substitusjonsprosessen ved tertiære karbonatomer, deretter ved sekundære karbonatomer, og til slutt ved primære karbonatomer.
Alkenes
Oksidasjonsprosesser avhenger av strukturen til alkenet og reaksjonsmiljøet.
1. Under oksidasjon av alkener med en konsentrert løsning av kaliumpermanganat KMnO 4 i et surt miljø (hard oksidasjon) σ- og π-bindinger brytes ved dannelse av karboksylsyrer, ketoner og karbonmonoksid (IV). Denne reaksjonen brukes til å bestemme posisjonen til dobbeltbindingen.
a) Hvis dobbeltbindingen er i enden av molekylet (for eksempel i buten-1), så er et av oksidasjonsproduktene maursyre, som lett oksideres til karbondioksid og vann:

b) Hvis karbonatomet ved dobbeltbindingen i et alkenmolekyl inneholder to karbonsubstituenter (for eksempel i molekylet 2-metylbuten-2), dannes det et keton under oksidasjonen., siden transformasjonen av et slikt atom til et atom av en karboksylgruppe er umulig uten å bryte CC-bindingen, som er relativt stabil under disse forholdene:

c) Hvis alkenmolekylet er symmetrisk og dobbeltbindingen er inneholdt i midten av molekylet, dannes det bare én syre under oksidasjon:

Et trekk ved oksidasjonen av alkener, der karbonatomene ved dobbeltbindingen inneholder to karbonradikaler, er dannelsen av to ketoner:


2. I nøytrale eller svakt alkaliske medier er oksidasjon ledsaget av dannelse av dioler (toverdige alkoholer) , og hydroksylgrupper er festet til de karbonatomene som det var en dobbeltbinding mellom:

Under denne reaksjonen blir den fiolette fargen på den vandige løsningen av KMnO 4 misfarget. Derfor brukes den som kvalitativ reaksjon til alkener (Wagner-reaksjon).
3. Oksidasjon av alkener i nærvær av palladiumsalter (Wacker-prosessen) fører til dannelsen aldehyder og ketoner:
2CH2=CH2 + O2 PdCl2/H2O→ 2 CH3-CO-H
Homologer oksideres ved det mindre hydrogenerte karbonatomet:
CH3-CH2-CH=CH2 + 1/2O2 PdCl2/H2O→ CH 3 - CH 2 -CO-CH 3
Alkyner
Oksydasjonen av acetylen og dets homologer skjer avhengig av miljøet hvor prosessen foregår.
EN) I et surt miljø er oksidasjonsprosessen ledsaget av dannelsen av karboksylsyrer:

Reaksjonen brukes til å bestemme strukturen til alkyner basert på deres oksidasjonsprodukter:

I nøytrale og svakt alkaliske miljøer er oksidasjonen av acetylen ledsaget av dannelsen av de tilsvarende oksalatene (oksalsyresalter), og oksidasjonen av homologer er ledsaget av brudd på trippelbindingen og dannelsen av karboksylsyresalter:

For acetylen:
1) I et surt miljø:
H-C=C-H KMnO 4, H 2 SÅ 4 → HOOC-COOH (oksalsyre)
3CH≡CH +8KMnO 4 H 2 O→ 3KOOC-KOOK kaliumoksalat+8MnO 2 ↓+ 2KOH+ 2H 2 O
Arenaer
(benzen og dets homologer)
Når arener oksideres i et surt miljø, bør man forvente dannelse av syrer, og i et alkalisk miljø - salter.
Benzenhomologer med én sidekjede (uavhengig av lengden) oksideres av et sterkt oksidasjonsmiddel til benzosyre ved α-karbonatomet. Ved oppvarming oksideres benzenhomologer av kaliumpermanganat i et nøytralt miljø for å danne kaliumsalter av aromatiske syrer.
5C 6 H 5 –CH 3 + 6KMnO 4 + 9H 2 SO 4 = 5C 6 H 5 COOH + 6MnSO 4 + 3K 2 SO 4 + 14H 2 O,
5C 6 H 5 –C 2 H 5 + 12KMnO 4 + 18H 2 SO 4 = 5C 6 H 5 COOH + 5CO 2 + 12MnSO 4 + 6K 2 SO 4 + 28H 2 O,
C 6 H 5 –CH 3 + 2KMnO 4 = C 6 H 5 COOK + 2MnO 2 + KOH + H 2 O.
Vi understreker at hvis det er flere sidekjeder i et arenmolekyl, blir hver av dem i et surt miljø oksidert ved a-karbonatomet til en karboksylgruppe, noe som resulterer i dannelsen av polybasiske aromatiske syrer:

1) I et surt miljø:
C6H5-CH2-R KMnO 4, H 2 SÅ 4 → C6H5-COOH benzosyre+CO2
2) I et nøytralt eller alkalisk miljø:
C6H5-CH2-R KMnO4, H2O/(OH)→ C 6 H 5 -COOK + CO 2
3) Oksidasjon av benzenhomologer med kaliumpermanganat eller kaliumdikromat ved oppvarming:
C6H5-CH2-R KMnO 4, H 2 SÅ 4, t ˚ C→ C6H5-COOH benzosyre+ R-COOH
4) Oksidasjon av kumen med oksygen i nærvær av en katalysator (kumenmetode for å produsere fenol):
C 6 H 5 CH(CH 3) 2 O2, H2SO4→C6H5-OH fenol + CH3-CO-CH3 aceton
5C 6 H 5 CH(CH 3) 2 + 18KMnO 4 + 27H 2 SO 4 → 5C 6 H 5 COOH + 42H 2 O + 18MnSO 4 + 10CO 2 + K 2 SO 4
C 6 H 5 CH(CH 3) 2 + 6H 2 O – 18ē→ C6H5COOH + 2CO2 + 18H + | x 5
MnO 4 - + 8H + + 5ē→ Mn +2 + 4H20 | x 18
Vær oppmerksom på at når mild oksidasjon av styren med kaliumpermanganat KMnO 4 i et nøytralt eller svakt alkalisk miljøπ-bindingen brytes og glykol (toverdig alkohol) dannes. Som et resultat av reaksjonen blir den fargede løsningen av kaliumpermanganat raskt misfarget og et brunt bunnfall av mangan (IV) oksid utfelles.
Oksidasjon sterkt oksidasjonsmiddel– kaliumpermanganat i et surt miljø – fører til fullstendig brudd på dobbeltbindingen og dannelse av karbondioksid og benzosyre, og løsningen blir misfarget.
C 6 H 5 −CH═CH 2 + 2 KMnO 4 + 3 H 2 SO 4 → C 6 H 5 −COOH + CO 2 + K 2 SO 4 + 2 MnSO 4 +4 H 2 O
Alkoholer
Det bør huskes at:
1) primære alkoholer oksideres til aldehyder:
3CH 3 –CH 2 OH + K 2 Cr 2 O 7 + 4H 2 SO 4 = 3CH 3 –CHO + K 2 SO 4 + Cr 2 (SO 4) 3 + 7H 2 O;
2) sekundære alkoholer oksideres til ketoner:

3) oksidasjonsreaksjon er ikke typisk for tertiære alkoholer.
Tertiære alkoholer, i molekylene der det ikke er noe hydrogenatom ved karbonatomet som inneholder OH-gruppen, oksiderer ikke under normale forhold. Under tøffe forhold (under påvirkning av sterke oksidasjonsmidler og ved høye temperaturer) kan de oksideres til en blanding av lavmolekylære karboksylsyrer, dvs. ødeleggelse av karbonskjelettet skjer.
Når metanol oksideres med en surgjort løsning av kaliumpermanganat eller kaliumdikromat, dannes CO 2.
Primære alkoholer under oksidasjon, avhengig av reaksjonsforholdene, kan danne ikke bare aldehyder, men også syrer.
For eksempel ender oksidasjonen av etanol med kaliumdikromat i kulden med dannelsen av eddiksyre, og ved oppvarming acetaldehyd:
3CH 3 –CH 2 OH + 2K 2 Cr 2 O 7 + 8H 2 SO 4 = 3CH 3 –COOH + 2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O,
Hvis tre eller flere OH-grupper er bundet til tilstøtende karbonatomer, blir midt- eller midtatomene omdannet til maursyre ved oksidasjon med perjodsyre
Oksydasjonen av glykoler med kaliumpermanganat i et surt miljø ligner på oksidativ spaltning av alkener og fører også til dannelse av syrer eller ketoner, avhengig av strukturen til den opprinnelige glykolen.
Aldehyder og ketoner
Aldehyder oksideres lettere enn alkoholer til de tilsvarende karboksylsyrene, ikke bare under påvirkning av sterke oksidanter (luftoksygen, surgjorte løsninger av KMnO 4 og K 2 Cr 2 O 7), men også under påvirkning av svake (ammoniakkløsning av sølvoksid eller kobber(II)hydroksid):
5CH 3 –CHO + 2KMnO 4 + 3H 2 SO 4 = 5CH 3 –COOH + 2MnSO 4 + K 2 SO 4 + 3H 2 O,
3CH 3 –CHO + K 2 Cr 2 O 7 + 4H 2 SO 4 = 3CH 3 –COOH + Cr 2 (SO 4) 3 + K 2 SO 4 + 4H 2 O,
CH 3 –CHO + 2OH CH 3 –COONH 4 + 2Ag + 3NH 3 + H 2 O
Spesiell oppmerksomhet!!! Oksidasjon av metanal med en ammoniakkløsning av sølvoksid fører til dannelse av ammoniumkarbonat i stedet for maursyre:
HCHOM+ 4OH = (NH 4) 2 CO 3 + 4Ag + 6NH 3 + 2H 2 O.
For å sette sammen ligninger for redoksreaksjoner brukes både elektronbalansemetoden og halvreaksjonsmetoden (elektronionmetoden).
For organisk kjemi er det ikke oksidasjonstilstanden til et atom som er viktig, men skiftet i elektrontetthet, som et resultat av at partielle ladninger vises på atomer som på ingen måte stemmer overens med verdiene til oksidasjonstilstander.
Mange universiteter inkluderer i opptaksbilletter oppgaver om å velge koeffisienter i OVR-ligninger ved bruk av den ione-elektroniske metoden (halvreaksjonsmetoden). Hvis i det minste en viss oppmerksomhet rettes mot denne metoden i skolen, er det hovedsakelig for oksidering av uorganiske stoffer.
La oss prøve å bruke halvreaksjonsmetoden for oksidasjon av sukrose med kaliumpermanganat i et surt miljø.
Fordelen med denne metoden er at det ikke er nødvendig å umiddelbart gjette og skrive ned reaksjonsproduktene. De bestemmes ganske enkelt av ligningen. Et oksidasjonsmiddel i et surt miljø viser sine oksiderende egenskaper best, for eksempel omdannes MnO - anion til Mn 2+ kation, lett oksiderte organiske forbindelser oksideres til CO 2.
La oss skrive ned transformasjonene av sukrose i molekylær form:
![]()
Det mangler 13 oksygenatomer på venstre side; for å eliminere denne motsetningen legger vi til 13 H 2 O-molekyler.
Den venstre siden inneholder nå 48 hydrogenatomer, de frigjøres i form av H + kationer:
La oss nå utjevne de totale ladningene på høyre og venstre side:
Halvreaksjonsordningen er klar. Å tegne et diagram over den andre halvreaksjonen forårsaker vanligvis ikke vanskeligheter:
La oss kombinere begge ordningene:
![]()
Oppdrag for selvstendig arbeid:
Fullfør CRM og ordne koeffisientene ved å bruke den elektroniske balansemetoden eller halvreaksjonsmetoden:
CH3-CH=CH-CH3 + KMnO4 + H2SO4 →
CH3-CH=CH-CH3 + KMnO4 + H2OM →
(CH 3) 2 C=C-CH 3 + KMnO 4 + H 2 SO 4 →
CH 3 -CH 2 -CH=CH 2 + KMnO 4 + H 2 SO 4 →
MEDH 3 -CH 2 -C≡C-CH 3 + KMnO 4 + H 2 SO 4 →
C6H5-CH3 + KMnO4 + H2O →
C 6 H 5 - C 2 H 5 + KMnO 4 + H 2 SO 4 →
C 6 H 5 - CH 3 + KMnO 4 + H 2 SÅ 4 →
Mine notater:
Elevene bør være spesielt oppmerksomme på oppførselen til oksidasjonsmidlet - kaliumpermanganat KMnO 4 i ulike miljøer. Dette skyldes det faktum at redoksreaksjoner i CMM-er ikke bare forekommer i oppgavene C1 og C2. I SZ-oppgaver som representerer en kjede av transformasjoner av organiske stoffer, er oksidasjons-reduksjonsligninger ikke uvanlige. På skolen er oksidasjonsmidlet ofte skrevet over pilen som [O]. Et krav for å fullføre slike oppgaver på Unified State Examination er obligatorisk utpeking av alle utgangsstoffer og reaksjonsprodukter med oppstilling av nødvendige koeffisienter.
I oppgaver i kategori C3 av Unified State Examination, er spesielle vanskeligheter forårsaket av oksidasjonsreaksjoner av organiske stoffer med kaliumpermanganat KMnO 4 i et surt miljø, som oppstår med brudd i karbonkjeden. For eksempel går oksidasjonsreaksjonen til propen i henhold til ligningen:
CH 3 – CH = CH 2 + KMnO4 + H 2 SÅ 4 → CH 3 COOH + CO 2 + MnSO 4 + K 2 SÅ 4 + H 2 O.
For å tilordne koeffisienter i komplekse ligninger av redoksreaksjoner som dette, foreslår standardteknikken å opprette en elektronisk balanse, men etter et nytt forsøk blir det åpenbart at dette ikke er nok. Roten til problemet her ligger i det faktum at koeffisienten før oksidasjonsmidlet, tatt fra den elektroniske balansen, må erstattes. Denne artikkelen tilbyr to metoder som lar deg velge riktig koeffisient før oksidasjonsmidlet for til slutt å utjevne alle elementene. Substitusjonsmetode for å erstatte koeffisienten før oksidasjonsmidlet, er det mer egnet for de som er i stand til å telle lenge og møysommelig, siden det kan være langvarig å ordne koeffisientene på denne måten (i dette eksemplet tok det 4 forsøk). Substitusjonsmetoden brukes sammen med "TABELL"-metoden, som også er omtalt i detalj i denne artikkelen. Den "algebraiske" metoden lar deg erstatte koeffisienten før oksidasjonsmidlet ikke mindre enkelt og pålitelig, men mye raskere KMnO4 Sammenlignet med substitusjonsmetoden har den imidlertid et snevrere anvendelsesområde. Den "algebraiske" metoden kan bare brukes til å erstatte koeffisienten før oksidasjonsmidlet KMnO4 i ligningene for oksidasjonsreaksjoner av organiske stoffer som oppstår ved brudd på karbonkjeden.
Nedlasting:
Forhåndsvisning:
For å bruke forhåndsvisningen, opprett en Google-konto og logg på den: https://accounts.google.com
Om temaet: metodologisk utvikling, presentasjoner og notater
Ordne koeffisienter i kjemiske ligninger
Læreren, som er hovedpersonen i å organisere den kognitive aktiviteten til elevene, leter stadig etter måter å forbedre effektiviteten av undervisningen. Organisering av effektiv trening...