I redoksreaksjoner organiske stoffer oftere viser de egenskapene til reduksjonsmidler, og selv blir oksidert. Den enkle oksidasjon av organiske forbindelser avhenger av tilgjengeligheten av elektroner når de interagerer med oksidasjonsmidlet. Alle kjente faktorer som forårsaker en økning i elektrontetthet i molekyler av organiske forbindelser (for eksempel positive induktive og mesomere effekter) vil øke deres evne til å oksidere og omvendt.
Tendensen til organiske forbindelser til å oksidere øker med deres nukleofilisitet, som tilsvarer følgende rader:
Økning i nukleofilisitet i serien
La oss vurdere redoksreaksjoner representanter for de viktigste klassene organisk materiale med noen uorganiske oksidasjonsmidler.
Oksidasjon av alkener
Ved mild oksidasjon omdannes alkener til glykoler (toverdige alkoholer). De reduserende atomene i disse reaksjonene er karbonatomer forbundet med en dobbeltbinding.
Reaksjonen med en løsning av kaliumpermanganat skjer i et nøytralt eller svakt alkalisk medium som følger:
3C 2 H 4 + 2KMnO 4 + 4H 2 O → 3CH 2 OH–CH 2 OH + 2MnO 2 + 2KOH
Under mer alvorlige forhold fører oksidasjon til brudd på karbonkjeden ved dobbeltbindingen og dannelse av to syrer (i et sterkt alkalisk miljø - to salter) eller en syre og karbondioksid (i et sterkt alkalisk miljø - et salt og et karbonat):
1) 5CH 3 CH=CHCH 2 CH 3 + 8KMnO 4 + 12H 2 SO 4 → 5CH 3 COOH + 5C 2 H 5 COOH + 8MnSO 4 + 4K 2 SO 4 + 17H 2 O
2) 5CH 3 CH=CH 2 + 10KMnO 4 + 15H 2 SO 4 → 5CH 3 COOH + 5CO 2 + 10MnSO 4 + 5K 2 SO 4 + 20H 2 O
3) CH 3 CH=CHCH 2 CH 3 + 8KMnO 4 + 10KOH → CH 3 COOK + C 2 H 5 COOK + 6H 2 O + 8K 2 MnO 4
4) CH 3 CH=CH 2 + 10KMnO 4 + 13KOH → CH 3 COOK + K 2 CO 3 + 8H 2 O + 10K 2 MnO 4
Kaliumdikromat i et svovelsyremedium oksiderer alkener på samme måte som reaksjon 1 og 2.
Under oksidasjonen av alkener, der karbonatomene ved dobbeltbindingen inneholder to karbonradikaler, dannes to ketoner:


Alkynoksidasjon
Alkyner oksiderer under litt mer alvorlige forhold enn alkener, så de oksiderer vanligvis ved å bryte karbonkjeden ved trippelbindingen. Som i tilfellet med alkener, er de reduserende atomene her karbonatomer forbundet med en multippelbinding. Som et resultat av reaksjonene dannes syrer og karbondioksid. Oksidasjon kan utføres med kaliumpermanganat eller dikromat i et surt miljø, for eksempel:
5CH 3 C≡CH + 8KMnO 4 + 12H 2 SO 4 → 5CH 3 COOH + 5CO 2 + 8MnSO 4 + 4K 2 SO 4 + 12H 2 O

Acetylen kan oksideres med kaliumpermanganat i et nøytralt miljø til kaliumoksalat:
3CH≡CH +8KMnO 4 → 3KOOC –COOK +8MnO 2 +2KOH +2H 2 O
I et surt miljø fortsetter oksidasjonen til oksalsyre eller karbondioksid:
5CH≡CH +8KMnO 4 +12H 2 SO 4 → 5HOOC –COOH +8MnSO 4 +4K 2 SO 4 +12H 2 O
CH≡CH + 2KMnO 4 +3H 2 SO 4 → 2CO 2 + 2MnSO 4 + 4H 2 O + K 2 SO 4
Oksidasjon av benzenhomologer
Benzen oksiderer ikke selv under ganske tøffe forhold. Benzenhomologer kan oksideres med en løsning av kaliumpermanganat i et nøytralt miljø til kaliumbenzoat:
C 6 H 5 CH 3 + 2KMnO 4 → C 6 H 5 COOK + 2MnO 2 + KOH + H 2 O
C 6 H 5 CH 2 CH 3 + 4KMnO 4 → C 6 H 5 COOK + K 2 CO 3 + 2H 2 O + 4MnO 2 + KOH
Oksidasjon av benzenhomologer med kaliumdikromat eller permanganat i et surt miljø fører til dannelse av benzosyre.
5C 6 H 5 CH 3 + 6KMnO 4 + 9 H 2 SO 4 → 5C 6 H 5 COOH+6MnSO 4 +3K 2 SO 4 + 14H 2 O
5C 6 H 5 –C 2 H 5 + 12KMnO 4 + 18H 2 SO 4 → 5C 6 H 5 COOH + 5CO 2 + 12MnSO 4 + 6K 2 SO 4 + 28H 2 O



Oksidasjon av alkoholer
Det direkte oksidasjonsproduktet av primære alkoholer er aldehyder, og oksidasjonsproduktene til sekundære alkoholer er ketoner.
Aldehyder dannet under oksidasjon av alkoholer oksideres lett til syrer, derfor oppnås aldehyder fra primære alkoholer ved oksidasjon med kaliumdikromat i et surt medium ved aldehydets kokepunkt. Når aldehyder fordamper, har de ikke tid til å oksidere.
3C 2 H 5 OH + K 2 Cr 2 O 7 + 4H 2 SO 4 → 3CH 3 CHO + K 2 SO 4 + Cr 2 (SO 4) 3 + 7H 2 O
Med et overskudd av oksidasjonsmiddel (KMnO 4, K 2 Cr 2 O 7) i ethvert miljø, oksideres primære alkoholer til karboksylsyrer eller deres salter, og sekundære alkoholer oksideres til ketoner.
5C 2 H 5 OH + 4KMnO 4 + 6H 2 SO 4 → 5CH 3 COOH + 4MnSO 4 + 2K 2 SO 4 + 11H 2 O
3CH 3 –CH 2 OH + 2K 2 Cr 2 O 7 + 8H 2 SO 4 → 3CH 3 –COOH + 2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O
Tertiære alkoholer oksiderer ikke under disse forholdene, men metylalkohol oksideres til karbondioksid.

Toverdig alkohol, etylenglykol HOCH 2 –CH 2 OH, ved oppvarming i et surt miljø med en løsning av KMnO 4 eller K 2 Cr 2 O 7, oksideres lett til oksalsyre, og i et nøytralt miljø til kaliumoksalat.
5CH 2 (OH) – CH 2 (OH) + 8КMnO 4 +12H 2 SO 4 → 5HOOC –COOH +8MnSO 4 +4К 2 SO 4 +22Н 2 О
3CH 2 (OH) – CH 2 (OH) + 8KMnO 4 → 3KOOC –COOK +8MnO 2 +2KOH +8H 2 O
Oksidasjon av aldehyder og ketoner
Aldehyder er ganske sterke reduksjonsmidler, og oksideres derfor lett av forskjellige oksidasjonsmidler, for eksempel: KMnO 4, K 2 Cr 2 O 7, OH, Cu(OH) 2. Alle reaksjoner oppstår ved oppvarming:
3CH 3 CHO + 2KMnO 4 → CH 3 COOH + 2CH 3 COOK + 2MnO 2 + H 2 O
3CH 3 CHO + K 2 Cr 2 O 7 + 4H 2 SO 4 → 3CH 3 COOH + Cr 2 (SO 4) 3 + 7H 2 O
CH 3 CHO + 2KMnO 4 + 3KOH → CH 3 COOK + 2K 2 MnO 4 + 2H 2 O
5CH 3 CHO + 2KMnO 4 + 3H 2 SO 4 → 5CH 3 COOH + 2MnSO 4 + K 2 SO 4 + 3H 2 O
CH 3 CHO + Br 2 + 3 NaOH → CH 3 COONa + 2 NaBr + 2H 2 O

"sølvspeil"-reaksjon
Med en ammoniakkløsning av sølvoksid oksideres aldehyder til karboksylsyrer, som i en ammoniakkløsning gir ammoniumsalter (“sølvspeil”-reaksjonen):
CH 3 CH=O + 2OH → CH 3 COONH 4 + 2Ag + H 2 O + 3NH 3
CH 3 –CH=O + 2Cu(OH) 2 → CH 3 COOH + Cu 2 O + 2H 2 O
Mauraldehyd (formaldehyd) oksideres vanligvis til karbondioksid:
5HCOH + 4KMnO4 (hytte) + 6H 2 SO 4 → 4MnSO 4 + 2K 2 SO 4 + 5CO 2 + 11 H 2 O
3CH 2 O + 2K 2 Cr 2 O 7 + 8H 2 SO 4 → 3CO 2 + 2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O
HCHO + 4OH → (NH 4) 2 CO 3 + 4Ag↓ + 2H 2 O + 6NH 3
HCOH + 4Cu(OH) 2 → CO 2 + 2Cu 2 O↓+ 5H 2 O
Ketoner oksideres under tøffe forhold av sterke oksidasjonsmidler med brudd på C-C-bindinger og gir blandinger av syrer:

Karboksylsyrer. Blant syrene har maursyre og oksalsyre sterke reduserende egenskaper, som oksiderer til karbondioksid.
HCOOH + HgCl2 =CO 2 + Hg + 2HCl
HCOOH+ Cl2 = CO2 +2HCl
HOOC-COOH+ Cl2 =2CO2 +2HCl
maursyre, i tillegg til sure egenskaper, viser også noen egenskaper til aldehyder, spesielt reduserende. Samtidig oksideres det til karbondioksid. For eksempel:
2KMnO4 + 5HCOOH + 3H2SO4 → K2SO4 + 2MnSO4 + 5CO2 + 8H2O
Ved oppvarming med sterke avvanningsmidler (H2SO4 (kons.) eller P4O10) spaltes det:
HCOOH →(t)CO + H2O
Katalytisk oksidasjon av alkaner:

Katalytisk oksidasjon av alkener:

Oksidasjon av fenoler:


Alkyner med en ikke-terminal trippelbinding tjener som en potensiell kilde for syntese av 1,2-diketoner under påvirkning av et passende oksidasjonsmiddel. Imidlertid er det ennå ikke funnet et universalreagens som forårsaker oksidasjon av en trippel karbon-karbonbinding til en 1,2-dikarbonylgruppe. RuO 4, ruthenium (VIII) oksid, foreslått for dette formålet er for dyrt og forårsaker ofte ytterligere oksidativ ødeleggelse av 1,2-diketoner til karboksylsyrer. Når disubstituerte acetylener interagerer med så sterke oksidasjonsmidler som kaliumpermanganat, er det bare i et helt nøytralt miljø ved pH 7–8 ved 0 °C som kan stoppes ved dannelsen av α-diketon. For eksempel blir stearolsyre ved pH 7,5 oksidert til α-diketon. I de fleste tilfeller er oksidasjon ledsaget av spaltning av trippelbindingen for å danne karboksylsyrer:

Utbyttet av produkter fra oksidativ destruksjon av alkyner er lite, og denne reaksjonen spiller ikke en vesentlig rolle i organisk syntese. Den brukes utelukkende for å bevise strukturen til naturlig forekommende acetylensyre som finnes i bladene til tropiske planter i Mellom-Amerika. Under dens oksidative ødeleggelse ble to syrer isolert - laurinsyre og adipin. Dette betyr at modersyren er 6-oktadecynsyre med et normalt karbonskjelett med sytten karbonatomer:
Mye viktigere er den oksidative koblingen av 1-alkyner katalysert av kobbersalter (Glaser-Eglinton-reaksjon). I 1870 oppdaget Glaser at en suspensjon av kobber (I) acetylid i alkohol oksideres av atmosfærisk oksygen for å danne 1,3-diyner:

For oksidasjon av kobber(I)acetylenider er kaliumheksacyanoferrat(III)K3 i DME eller DMF mer effektivt som oksidasjonsmiddel. I 1959 foreslo Eglinton en mye mer praktisk modifikasjon av den oksidative kondensasjonen av alkyner. Alkyn oksideres med kobber(II)acetat i en pyridinløsning ved 60–70 °C. Eglinton-modifikasjonen har vist seg å være ekstremt nyttig for syntese av makrosykliske polyyner fra ,-diynes. Som en illustrasjon presenterer vi syntesen av to syklopolyiner under den oksidative kondensasjonen av heksadiin-1,5 (F. Sondheimer, 1960):

En av polyinene er et produkt av cyklotrimerisering, den andre er et produkt av cyklotetramerisering av det opprinnelige hesadiin-1,5. Trimeren fungerer som startreagens for syntesen av aromatisk -annulen (for mer informasjon om annulener, se kapittel 12). På samme måte, under de samme forholdene som nonadiin-1,8, oppnås dens dimer - 1,3,10,12-cyclooctadecatetraene, sammen med en trimer, tetramer og pentamer:

For å oppnå usymmetriske diyner brukes kondensering av haloacetylener med alkyn-1 (terminal alkyn) i nærvær av kobber (I) salter og et primært amin (kombinasjon i henhold til Kadio-Khodkevich, 1957):

Utgangsbromalkynene oppnås ved virkningen av natriumhypobromitt på alkyner-1 eller fra litium- og bromacetylenider:
Organokobberderivatet av det terminale alkynet genereres direkte i reaksjonsblandingen av Cu 2 Cl 2 og alkyn-1.
6.3.4. Elektrofile addisjonsreaksjoner til en trippelbinding
Elektrofile addisjonsreaksjoner til en trippelbinding er blant de mest typiske og viktige reaksjonene til alkyner. I motsetning til elektrofil tilsetning til alkener, var den syntetiske anvendelsen av denne store gruppen av reaksjoner langt foran utviklingen av teoretiske ideer om dens mekanisme. Men i løpet av de siste tjue årene har situasjonen endret seg betydelig, og for tiden er dette et av de raskt utviklende områdene innen fysisk organisk kjemi. HOMO-en til en alkyn ligger lavere enn HOMO-en til en alken (kapittel 2), og denne omstendigheten bestemmer i de aller fleste tilfeller en lavere tilsetningshastighet av et elektrofilt middel til en alkyn sammenlignet med en alken. En annen faktor som bestemmer forskjellen i reaktiviteten til alkyner og alkener i elektrofile addisjonsreaksjoner er den relative stabiliteten til mellomproduktene som følge av tilsetning av en elektrofil art til trippel- og dobbeltbindinger. Når en elektrofil H + eller E + art fester seg til en dobbeltbinding, dannes en syklisk eller åpen karbokation (kapittel 5). Tilsetning av H+ eller E+ til en trippelbinding resulterer i dannelsen av et åpent eller syklisk vinylkation. I en lineær åpen vinylkation er det sentrale karbonatomet lokalisert i sp-hybrid tilstand, mens den er ledig R-orbital er ortogonal til -bindingen. Fordi det sp-hybridkarbonatomet til vinylkationen har en høyere elektronegativitet sammenlignet med sp 2-hybridatomet til alkylkationen, bør vinylkationen være mindre stabil sammenlignet med alkylkationen:

Data fra kvantemekaniske beregninger, samt termodynamiske data for gassfasen oppnådd ved bruk av høytrykks massespektrometri og syklotronresonansspektroskopi, er i full overensstemmelse med disse betraktningene. I tabellen 6.3 viser termodynamiske data for dannelse av en rekke karbokasjoner og hydrokarboner relatert til gassfasen ved 25 °C.
|
Karbokasjon |
Δ N f ˚ kcal/mol |
|
| |
|
| |
|
|
Fra dataene presentert i tabellen. 6.3, følger det at vinylkationen er 47 kcal/mol mindre stabil enn etylkationen som inneholder samme antall atomer. Den samme konklusjonen kan trekkes fra ioniseringsentalpien i gassfasen CH 3 CH 2 Cl og CH 2 = CHCl:
Det er lett å se at kombinasjonen av begge faktorene - den høyere energien til vinylkationen og den lavtliggende HOMO av alkynen - representerer en lavere reaktivitet av alkyner sammenlignet med alkener i elektrofile addisjonsreaksjoner. I tabellen 6.4 inneholder sammenlignende data om tilsetning av halogener, sulfen- og selenylklorider, trifluoreddiksyre og vann til forskjellige alkener og alkyner som ikke inneholder noen aktiverende eller deaktiverende funksjonell gruppe.
Tabell 6.4
Sammenlignende egenskaper for alkyner og alkener
i elektrofile addisjonsreaksjoner
|
Underlag |
K alken/K alkyn |
|
|
Bromering i eddiksyre |
CH 2 CH 2 /HCCH C 4 H 9 CH = CH 2 / C 4 H 9 CCH C 6 H 5 CH = CH 2 / C 6 H 5 CCH | |
|
Klorering i eddiksyre |
C 6 H 5 CH = CH 2 / C 6 H 5 CCH C 4 H 9 CH=CH 2 /C 6 H 5 CCH С 2 Н 5 С=СНС 2 Н 5 /С 2 Н 5 ССС 2 Н 5 | |
|
Tilsetning av 4-klorfenylsulfenklorid P-ClС 6 H 4 SeCl |
CH 2 =CH 2 /HCCH C 4 H 9 CH = CH 2 / C 4 H 9 CCH C 6 H 5 CH = CH 2 / C 6 H 5 CCH | |
|
Tilsetning av fenylselenklorid C 6 H 5 SeCl |
CH 2 =CH 2 /HCCH C 4 H 9 CH = CH 2 / C 4 H 9 CCH C 6 H 5 CH = CH 2 / C 6 H 5 CCH | |
|
Tilsetning av trifluoreddiksyre |
C 4 H 9 CH = CH 2 / C 4 H 9 CCH C 6 H 5 CH = CH 2 / C 6 H 5 CCH C 2 H 5 CH = CH 2 / C 2 H 5 CCH | |
|
Syrekatalysert hydrering |
C 4 H 9 CH = CH 2 / C 4 H 9 CCH С 2 Н 5 СН=СНС 2 Н 5 /С 2 Н 5 ССС 2 Н 5 C 6 H 5 CH = CH 2 / C 6 H 5 CCH |
Fra disse dataene følger det at bare tilsetning av sure midler og vann til trippel- og dobbeltbindinger skjer med lignende hastigheter. Tilsetningen av halogener, sulfenklorider og en rekke andre reagenser til alkener skjer 10 2 - 10 5 ganger raskere enn til alkyner. Dette betyr at hydrokarboner som inneholder ikke-konjugerte trippel- og dobbeltbindinger selektivt tilsetter disse reagensene ved dobbeltbindingen, for eksempel:

Data om komparativ hydrering av alkyner og alkener bør behandles med forsiktighet fordi hydrering av alkyner krever kvikksølv(II)ionkatalyse, som er ineffektiv for tilsetning av vann til dobbeltbindingen. Derfor er data om hydrering av trippel- og dobbeltbindinger strengt tatt ikke sammenlignbare.
Tilsetningen av halogener, hydrogenhalogenider, sulfenklorider og andre elektrofile midler kan utføres i trinn, som lett illustreres ved hjelp av følgende eksempler:



Tendensen til organiske forbindelser til å oksidere er assosiert med tilstedeværelsen av flere bindinger, funksjonelle grupper og hydrogenatomer ved karbonatomet som inneholder den funksjonelle gruppen. Den sekvensielle oksidasjonen av organiske stoffer kan representeres som følgende kjede av transformasjoner:
Mettet hydrokarbon → Umettet hydrokarbon → Alkohol → Aldehyd (keton) → Karboksylsyre → CO2 + H2O
Det genetiske forholdet mellom klasser av organiske forbindelser er her representert som en rekke redoksreaksjoner som sikrer overgangen fra en klasse av organiske forbindelser til en annen. Det fullføres av produktene av fullstendig oksidasjon (forbrenning) av enhver representant for klassene av organiske forbindelser. Avhengighet av redoksevnen til et organisk stoff på dets struktur: Den økte tendensen til organiske forbindelser til å oksidere skyldes tilstedeværelsen av stoffer i molekylet: flere bindinger (det er derfor alkener, alkyner, alkadiener oksideres så lett); visse funksjonelle grupper som lett kan oksideres (–-SH, –OH (fenoliske og alkoholiske), – NH2; aktiverte alkylgrupper lokalisert ved siden av flere bindinger.
For eksempel kan propen oksideres til det umettede aldehyd akrolein med atmosfærisk oksygen i nærvær av vanndamp på vismut-molybden-katalysatorer.
H2C═CH−CH3 → H2C═CH−COH
Og også oksidasjon av toluen til benzosyre med kaliumpermanganat i et surt miljø. 5C6H5CH3 +6KMnO4 + 9H2SO4 → 5C6H5COOH + 3K2SO4 + 6MnSO4 +14H2O
tilstedeværelsen av hydrogenatomer ved et karbonatom som inneholder en funksjonell gruppe. Et eksempel er reaktiviteten i oksidasjonsreaksjonene til primære, sekundære og tertiære alkoholer ved oksidasjonsreaktivitet.
Til tross for at det under enhver redoksreaksjon oppstår både oksidasjon og reduksjon, klassifiseres reaksjoner avhengig av hva som skjer direkte med den organiske forbindelsen (hvis den er oksidert, snakker vi om oksidasjonsprosessen, hvis den reduseres, snakker vi om reduksjonsprosessen ).
Således, i reaksjonen av etylen med kaliumpermanganat, vil etylen oksideres, og kaliumpermanganat reduseres. Reaksjonen kalles etylenoksidasjon.
Når vi studerte de komparative egenskapene til uorganiske og organiske forbindelser, ble vi kjent med bruken av oksidasjonstilstand (s.o.) (i organisk kjemi, først og fremst karbon) og metoder for å bestemme den:
1) beregning av gjennomsnitt s.o. karbon i et molekyl av organisk materiale: -8/3 +1 C3 H8 Denne tilnærmingen er berettiget hvis under reaksjonen i det organiske stoffet blir alle kjemiske bindinger ødelagt (forbrenning, fullstendig nedbrytning).
2) definisjon av s.o. hvert karbonatom:
I dette tilfellet er oksidasjonstilstanden til ethvert karbonatom i en organisk forbindelse lik den algebraiske summen av tallene for alle bindinger med atomer av mer elektronegative elementer, tatt i betraktning med "+"-tegnet på karbonatomet, og antall bindinger med hydrogenatomer (eller et annet mer elektropositivt element), tatt i betraktning med tegnet "-" ved karbonatomet. I dette tilfellet tas det ikke hensyn til bindinger med nabokarbonatomer. Som et enkelt eksempel, la oss bestemme oksidasjonstilstanden til karbon i et metanolmolekyl. Et karbonatom er koblet til tre hydrogenatomer (disse bindingene telles med et "–"-tegn), og en binding er koblet til et oksygenatom (det telles med et "+"-tegn). Vi får: -3 + 1 = -2. Dermed er oksidasjonstilstanden til karbon i metanol -2. Den beregnede graden av oksidasjon av karbon, selv om den er en betinget verdi, indikerer arten av skiftet i elektrontetthet i molekylet, og endringen som et resultat av reaksjonen indikerer at redoksprosessen finner sted. La oss avklare i hvilke tilfeller det er bedre å bruke en eller annen metode.
Prosessene med oksidasjon, forbrenning, halogenering, nitrering, dehydrogenering og dekomponering er klassifisert som redoksprosesser. Når man flytter fra en klasse av organiske forbindelser til en annen og øker graden av forgrening av karbonskjelettet til molekylene av forbindelser innenfor en egen klasse, endres graden av oksidasjon av karbonatomet som er ansvarlig for den reduserende evnen til forbindelsen. Organiske stoffer, hvis molekyler inneholder karbonatomer med maksimale (- og +) CO-verdier (-4, -3, +2, +3), inngår en fullstendig oksidasjons-forbrenningsreaksjon, men er motstandsdyktige mot milde og middels sterke oksidasjonsmidler. Stoffer hvis molekyler inneholder karbonatomer i CO-1; 0; +1, oksider lett, deres reduserende evner er nære, så deres ufullstendige oksidasjon kan oppnås ved å bruke et av de kjente oksidasjonsmidlene med lav og middels styrke. Disse stoffene kan ha en dobbel natur, og fungere som et oksidasjonsmiddel, akkurat som det er iboende i uorganiske stoffer.
Alkaner

Alkenes
Oksidasjonsprosesser avhenger av strukturen til alkenet og reaksjonsmiljøet.

1. Når alkener oksideres med en konsentrert løsning av kaliumpermanganat KMnO4 i et surt miljø (hard oksidasjon), brytes σ- og π-bindinger for å danne karboksylsyrer, ketoner og karbonmonoksid (IV). Denne reaksjonen brukes til å bestemme posisjonen til dobbeltbindingen.

a) Hvis dobbeltbindingen er i enden av molekylet (for eksempel i buten-1), så er et av oksidasjonsproduktene maursyre, som lett oksideres til karbondioksid og vann:

b) Hvis karbonatomet ved dobbeltbindingen i et alkenmolekyl inneholder to karbonsubstituenter (for eksempel i 2-metylbuten-2 molekylet), dannes det under oksidasjonen et keton, siden transformasjonen av et slikt atom inn i et karboksylgruppeatom er umulig uten å bryte C-C-binding, relativt stabil under disse forholdene:

c) Hvis alkenmolekylet er symmetrisk og dobbeltbindingen er inneholdt i midten av molekylet, dannes det bare én syre under oksidasjon:

Et trekk ved oksidasjonen av alkener, der karbonatomene ved dobbeltbindingen inneholder to karbonradikaler, er dannelsen av to ketoner:


2. I nøytrale eller svakt alkaliske medier er oksidasjon ledsaget av dannelsen av dioler (toverdige alkoholer), og hydroksylgrupper tilsettes de karbonatomene som det var en dobbeltbinding mellom:

Under denne reaksjonen blir den fiolette fargen til den vandige KMnO4-løsningen misfarget. Derfor brukes den som en kvalitativ reaksjon for alkener (Wagner-reaksjon).
3. Oksidasjon av alkener i nærvær av palladiumsalter (Wacker-prosessen) fører til dannelse av aldehyder og ketoner:
2CH2=CH2 + O2 PdCl2/H2O → 2 CH3-CO-H
Homologer oksideres ved det mindre hydrogenerte karbonatomet: CH3-CH2-CH=CH2 + 1/2O2 PdCl2/H2O → CH3-CH2-CO-CH3 Alkyner
Oksydasjonen av acetylen og dets homologer skjer avhengig av miljøet hvor prosessen foregår.
a) I et surt miljø er oksidasjonsprosessen ledsaget av dannelsen av karboksylsyrer:

1 Reaksjonen brukes til å bestemme strukturen til alkyner basert på deres oksidasjonsprodukter:

2 I nøytrale og svakt alkaliske miljøer er oksidasjonen av acetylen ledsaget av dannelsen av de tilsvarende oksalater (oksalsyresalter), og oksidasjonen av homologer er ledsaget av brudd på trippelbindingen og dannelse av karboksylsyresalter:

3 For acetylen:
1) I et surt miljø: H-C≡C-H KMnO4, H2SO4→ HOOC-COOH (oksalsyre)
2) I et nøytralt eller alkalisk miljø: 3CH≡CH +8KMnO4 H2O→ 3KOOC-COOK kaliumoksalat +8MnO2↓+ 2KOH+ 2H2O
Arenaer (benzen og dets homologer)
Når arener oksideres i et surt miljø, bør man forvente dannelse av syrer, og i et alkalisk miljø - salter. Benzenhomologer med én sidekjede (uavhengig av lengden) oksideres av et sterkt oksidasjonsmiddel til benzosyre ved α-karbonatomet. Ved oppvarming oksideres benzenhomologer av kaliumpermanganat i et nøytralt miljø for å danne kaliumsalter av aromatiske syrer.
5C6H5–CH3 + 6KMnO4 + 9H2SO4 = 5C6H5COOH + 6MnSO4 + 3K2SO4 + 14H2O,
5C6H5–C2H5 + 12KMnO4 + 18H2SO4 = 5C6H5COOH + 5CO2 + 12MnSO4 + 6K2SO4 + 28H2O,
C6H5–CH3 + 2KMnO4 = C6H5COOK + 2MnO2 + KOH + H2O.
Vi understreker at hvis det er flere sidekjeder i et arenmolekyl, blir hver av dem i et surt miljø oksidert ved a-karbonatomet til en karboksylgruppe, noe som resulterer i dannelsen av polybasiske aromatiske syrer:

1) I et surt miljø: C6H5-CH2-R KMnO4, H2SO4→ C6H5-COOH benzosyre + CO2
2) I et nøytralt eller alkalisk miljø: C6H5-CH2-R KMnO4, H2O/(OH)→ C6H5-COOK + CO2
3) Oksidasjon av benzenhomologer med kaliumpermanganat eller kaliumdikromat ved oppvarming: C6H5-CH2-R KMnO4, H2SO4, t˚C→ C6H5-COOHbenzosyre + R-COOH
4) Oksidasjon av kumen med oksygen i nærvær av en katalysator (kumenmetode for å produsere fenol): C6H5CH(CH3)2 O2, H2SO4→ C6H5-OH fenol + CH3-CO-CH3 aceton
5C6H5CH(CH3)2 + 18KMnO4 + 27H2SO4 → 5C6H5COOH + 42H2O + 18MnSO4 + 10CO2 + K2SO4
Det skal bemerkes at under mild oksidasjon av styren med kaliumpermanganat KMnO4 i et nøytralt eller svakt alkalisk medium, brytes π-bindingen og glykol (toverdig alkohol) dannes. Som et resultat av reaksjonen blir den fargede løsningen av kaliumpermanganat raskt misfarget og et brunt bunnfall av mangan (IV) oksid utfelles. Oksidasjon med et sterkt oksidasjonsmiddel - kaliumpermanganat i et surt miljø - fører til fullstendig brudd på dobbeltbindingen og dannelse av karbondioksid og benzosyre, og løsningen blir misfarget.
C6H5−CH═CH2 + 2 KMnO4 + 3 H2SO4 → C6H5−COOH + CO2 + K2SO4 + 2 MnSO4 +4 H2O
Alkoholer
Det bør huskes at:
1) primære alkoholer oksideres til aldehyder: 3CH3–CH2OH + K2Cr2O7 + 4H2SO4 = 3CH3–CHO + K2SO4 + Cr2(SO4)3 + 7H2O;
2) sekundære alkoholer oksideres til ketoner:

3) oksidasjonsreaksjon er ikke typisk for tertiære alkoholer. Tertiære alkoholer, i molekylene der det ikke er noe hydrogenatom ved karbonatomet som inneholder OH-gruppen, oksiderer ikke under normale forhold. Under tøffe forhold (under påvirkning av sterke oksidasjonsmidler og ved høye temperaturer) kan de oksideres til en blanding av lavmolekylære karboksylsyrer, dvs. ødeleggelse av karbonskjelettet skjer. Når metanol oksideres med en surgjort løsning av kaliumpermanganat eller kaliumdikromat, dannes CO2. Primære alkoholer under oksidasjon, avhengig av reaksjonsforholdene, kan danne ikke bare aldehyder, men også syrer. For eksempel ender oksidasjonen av etanol med kaliumdikromat i kulden med dannelsen av eddiksyre, og ved oppvarming acetaldehyd:
3CH3–CH2OH + 2K2Cr2O7 + 8H2SO4 = 3CH3–COOH + 2K2SO4 + 2Cr2(SO4)3 + 11H2O,
3CH3–CH2OH + K2Cr2O7 + 4H2SO4
3CH3–CHO + K2SO4 + Cr2(SO4)3 + 7H2O
Vi husker påvirkningen av miljøet på produktene av alkoholoksidasjonsreaksjoner, nemlig: en varm nøytral løsning av KMnO4 oksiderer metanol til kaliumkarbonat, og de resterende alkoholene til salter av de tilsvarende karboksylsyrene:

Oksidasjon av glykoler
1,2-glykoler spaltes lett under milde forhold ved innvirkning av periodic acid. Avhengig av strukturen til den opprinnelige glykolen, kan oksidasjonsprodukter være aldehyder eller ketoner:
![]()
Hvis tre eller flere OH-grupper er bundet til tilstøtende karbonatomer, blir midt- eller midtatomene omdannet til maursyre ved oksidasjon med perjodsyre
Oksydasjonen av glykoler med kaliumpermanganat i et surt miljø ligner på oksidativ spaltning av alkener og fører også til dannelse av syrer eller ketoner, avhengig av strukturen til den opprinnelige glykolen.
Aldehyder og ketoner
Aldehyder oksideres lettere enn alkoholer til de tilsvarende karboksylsyrene, ikke bare under påvirkning av sterke oksidanter (luftoksygen, surgjorte løsninger av KMnO4 og K2Cr2O7), men også under påvirkning av svake (ammoniakkløsning av sølvoksid eller kobber(II) ) hydroksid):
5CH3–CHO + 2KMnO4 + 3H2SO4 = 5CH3–COOH + 2MnSO4 + K2SO4 + 3H2O,
3CH3–CHO + K2Cr2O7 + 4H2SO4 = 3CH3–COOH + Cr2(SO4)3 + K2SO4 + 4H2O,
CH3–CHO + 2OH CH3–COONH4 + 2Ag + 3NH3 + H2O
Spesiell oppmerksomhet!!! Oksidasjon av metanal med en ammoniakkløsning av sølvoksid fører til dannelse av ammoniumkarbonat i stedet for maursyre: HCHO + 4OH = (NH4)2CO3 + 4Ag + 6NH3 + 2H2O.
Som allerede nevnt, er oksidasjonen av et organisk stoff innføring av oksygen i dets sammensetning og (eller) eliminering av hydrogen. Reduksjon er omvendt prosess (introduksjon av hydrogen og eliminering av oksygen). Med tanke på sammensetningen av alkaner (СnH2n+2), kan vi konkludere med at de ikke er i stand til å delta i reduksjonsreaksjoner, men kan delta i oksidasjonsreaksjoner.
Alkaner er forbindelser med lave oksidasjonstilstander av karbon, og avhengig av reaksjonsforholdene kan de oksideres til forskjellige forbindelser.
Ved vanlige temperaturer reagerer ikke alkaner selv med sterke oksidasjonsmidler (H2Cr2O7, KMnO4, etc.). Når de introduseres i åpen flamme, brenner alkaner. I dette tilfellet, i et overskudd av oksygen, skjer deres fullstendige oksidasjon til CO2, der karbon har den høyeste oksidasjonstilstanden på +4, og vann. Forbrenningen av hydrokarboner fører til brudd på alle C-C og CH-bindinger og er ledsaget av frigjøring av en stor mengde varme (eksoterm reaksjon).
Det er generelt akseptert at mekanismen for oksidasjon av alkaner involverer en radikalkjedeprosess, siden oksygen i seg selv er dårlig reaktivt; for å abstrahere et hydrogenatom fra en alkan, trengs en partikkel som vil sette i gang dannelsen av et alkylradikal, som vil reagere med oksygen og gi et peroksyradikal. Peroksyradikalet kan deretter abstrahere et hydrogenatom fra et annet alkanmolekyl for å danne et alkylhydroperoksid og radikal.

Det er mulig å oksidere alkaner med atmosfærisk oksygen ved 100-150°C i nærvær av en katalysator - manganacetat; denne reaksjonen brukes i industrien. Oksidasjon oppstår når en strøm av luft blåses gjennom smeltet parafin som inneholder et mangansalt.
Fordi Som et resultat av reaksjonen dannes en blanding av syrer, de separeres fra ureagert parafin ved å oppløses i vandig alkali, og deretter nøytraliseres med mineralsyre.
Direkte i industrien brukes denne metoden for å få eddiksyre fra n-butan:
Oksidasjon av alkener
Oksidasjonsreaksjoner av alkener er delt inn i to grupper: 1) reaksjoner der karbonskjelettet er bevart, 2) reaksjoner med oksidativ ødeleggelse av karbonskjelettet til molekylet ved dobbeltbindingen.
Oksidasjonsreaksjoner av alkener med bevaring av karbonskjelettet
1. Epoksidasjon (Prilezhaev-reaksjon)
Asykliske og sykliske alkener, når de reagerer med persyrer i et ikke-polart miljø, danner epoksider (oksiraner).
Oksiraner kan også oppnås ved oksidasjon av alkener med hydroperoksider i nærvær av molybden-, wolfram- og vanadiumholdige katalysatorer:

Det enkleste oksiranet, etylenoksid, produseres industrielt ved oksidasjon av etylen med oksygen i nærvær av sølv eller sølvoksid som katalysator.

2. anti-hydroksylering (hydrolyse av epoksider)
Sur (eller alkalisk) hydrolyse av epoksider fører til åpning av oksidringen med dannelse av transdioler.

I det første trinnet protoneres oksygenatomet til epoksidet for å danne et syklisk oksoniumkation, som åpner seg som et resultat av det nukleofile angrepet av vannmolekylet.
Basekatalysert epoksyringåpning fører også til dannelse av transglykoler.

3. syn-hydroksylering
En av de eldste metodene for oksidasjon av alkener er Wagner-reaksjonen (oksidasjon med kaliumpermanganat). Opprinnelig produserer oksidasjon en syklisk ester av mangansyre, som hydrolyserer til en nærliggende diol:

I tillegg til Wagner-reaksjonen er det en annen metode for syn-hydroksylering av alkener under påvirkning av osmium (VIII) oksid, som ble foreslått av Krige. Når osmiumtetroksid reagerer med en alken i eter eller dioksan, dannes et svart bunnfall av syklisk osmisk syreester, osmat. Imidlertid akselereres tilsetningen av OsO4 til multippelbindingen merkbart i pyridin. Det resulterende sorte osmatutfellingen spaltes lett ved virkningen av en vandig løsning av natriumhydrosulfitt:

Kaliumpermanganat eller osmium(VIII)oksid oksiderer alkenet til cis-1,2-diol.
Oksidativ spaltning av alkener
Den oksidative spaltningen av alkener inkluderer reaksjoner av deres interaksjon med kaliumpermanganat i alkalisk eller svovelsyre, samt oksidasjon med en løsning av kromtrioksid i eddiksyre eller kaliumdikromat og svovelsyre. Sluttresultatet av slike transformasjoner er spaltningen av karbonskjelettet på stedet for dobbeltbindingen og dannelsen av karboksylsyrer eller ketoner.
Monosubstituerte alkener med en terminal dobbeltbinding spaltes til karboksylsyre og karbondioksid:

Hvis begge karbonatomene ved en dobbeltbinding inneholder bare en alkylgruppe, dannes en blanding av karboksylsyrer:

Men hvis alkenet tetrasubstituert ved dobbeltbindingen er en keton:

Reaksjonen av ozonolyse av alkener har en mye større preparativ betydning. I mange tiår fungerte denne reaksjonen som hovedmetoden for å bestemme strukturen til foreldrealkenen. Denne reaksjonen utføres ved å sende en strøm av en løsning av ozon i oksygen, en løsning av en alken i metylenklorid eller etylacetat ved -80 ... -100 ° C. Mekanismen for denne reaksjonen ble etablert av Krige:


Ozonider er ustabile forbindelser som spaltes eksplosivt. Det er to måter å spalte ozonider på - oksidativt og reduktivt.
Under hydrolyse brytes ozonider ned til karbonylforbindelser og hydrogenperoksid. Hydrogenperoksid oksiderer aldehyder til karboksylsyrer - dette er oksidativ nedbrytning:

Mye viktigere er den reduktive spaltningen av ozonider. Produktene fra ozonolyse er aldehyder eller ketoner, avhengig av strukturen til startalkenet:
I tillegg til metodene ovenfor, er det en annen metode foreslått i 1955 av Lemieux:
I Lemieux-metoden er det ingen arbeidskrevende prosedyrer for å separere mangandioksid, fordi dioksid og manganat oksideres igjen av perjodat til permanganation. Dette tillater bare å bruke katalytiske mengder kaliumpermanganat.
4.5. Oksidasjon av alkener
Det er tilrådelig å dele oksidasjonsreaksjonene til alkener i to store grupper: reaksjoner der karbonskjelettet er bevart og reaksjoner med oksidativ ødeleggelse av karbonskjelettet til molekylet ved dobbeltbindingen. Den første gruppen av reaksjoner inkluderer epoksidering, så vel som hydroksylering, som fører til dannelse av vicinale dioler (glykoler). Når det gjelder sykliske alkener, produserer hydroksylering vicinal transe- eller cis-dioler. En annen gruppe inkluderer ozonolyse og uttømmende oksidasjonsreaksjoner av alkener, som fører til dannelse av ulike typer karbonylforbindelser og karboksylsyrer.
4.5.a. Oksidasjonsreaksjoner av alkener med bevaring av karbonskjelettet
1. Epoksidasjon (reaksjon av N.A. Prilezhaev, 1909)
Asykliske og sykliske alkener, når de reagerer med persyrer (persyrer) RCOOOH i et ikke-polart, likegyldig miljø, danner epoksider (oksiraner), derfor kalles selve reaksjonen epoksidasjonsreaksjonen.
I følge moderne nomenklatur IUPAC- en treleddet ring med ett oksygenatom kalles oksiran.
Epoksidering av alkener bør betraktes som en synkron, koordinert prosess der ioniske mellomprodukter som hydroksylkationen OH+ ikke deltar. Med andre ord er epoksidering av alkener en prosess syn-binding av ett oksygenatom til en dobbeltbinding med fullstendig bevaring av konfigurasjonen av substituenter ved dobbeltbindingen.
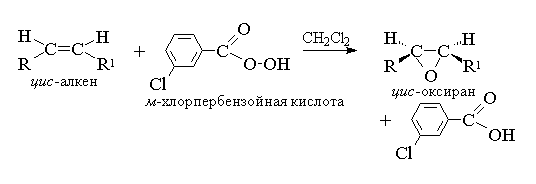

En mekanisme som er karakteristisk for samordnede prosesser har blitt foreslått for epoksidering.

Siden angrep av dobbeltbindingen av oksygenatomet til persyren er like sannsynlig på begge sider av dobbeltbindingsplanet, er de resulterende oksiranene enten meso-former eller blandinger av enantiomerer. Følgende persyrer brukes som epoksideringsmidler: perbenzoic, m-klorperbenzosyre, monoperftalsyre, pereddiksyre, trifluorpereddiksyre og performisk. Persyrer av den aromatiske serien brukes i form av individuelle reagenser, mens persyrer av den alifatiske serien - CH 3 CO 3 H, CF 3 CO 3 H og HCO 3 H ikke isoleres i individuell form, men brukes etter dannelse i interaksjonen mellom 30% eller 90% hydrogenperoksid og den tilsvarende karboksylsyren. Perbenzoin og m-klorperbenzosyre oppnås ved oksidasjon av benzosyre og m-klorbenzosyrer med 70% hydrogenperoksyd i en løsning av metansulfonsyre eller fra syrekloridene av disse syrene og hydrogenperoksyd.


Monoperftalsyre oppnås ved en lignende metode fra ftalsyreanhydrid og 30 % hydrogenperoksid.

Opprinnelig ble perbenzosyre eller monoperftalsyre brukt for å oppnå oksiraner (epoksider):

For tiden er den mest brukte for epoksidering m-klorperbenzosyre. I motsetning til andre persyrer er den holdbar i lang tid (opptil 1 år) og er helt sikker å håndtere. Utbytter av oksiraner oppnådd ved oksidasjon av asykliske og sykliske alkener m-klorperbenzosyre i en løsning av metylenklorid, kloroform eller dioksan er vanligvis ganske høye.



Persyrer genereres ofte direkte i en reaksjonsblanding av 90 % hydrogenperoksid og karboksylsyre i metylenklorid.


Alkener med en dobbeltbinding konjugert til en karbonylgruppe eller annen akseptorsubstituent er inaktive og for deres oksidasjon er det bedre å bruke sterkere oksidasjonsmidler, slik som trifluorpereddiksyre, oppnådd fra trifluoreddiksyreanhydrid og 90 % hydrogenperoksid i metylenklorid. Det enkleste oksiranet, etylenoksid, produseres industrielt ved oksidasjon av etylen med oksygen i nærvær av sølv som katalysator.

2. anti- Hydroksylering
Den tre-leddede ringen av oksiraner åpnes lett under påvirkning av en rekke nukleofile reagenser. Disse reaksjonene vil bli diskutert i detalj i avsnittet om asykliske og sykliske etere. Her vil bare hydrolyse av oksiraner bli vurdert. Hydrolysen av oksiraner katalyseres av både syrer og baser. I begge tilfeller dannes vicinale dioler, dvs. glykoler. I syrekatalyse, i det første trinnet, protoneres oksyran-oksygenatomet for å danne et syklisk oksoniumkation, som åpner seg som et resultat av det nukleofile angrepet av et vannmolekyl:

Nøkkeltrinnet i ringåpning, som bestemmer hastigheten på hele prosessen, er det nukleofile angrepet av vann på den protonerte formen av oksiran. Mekanistisk ligner denne prosessen åpningen av et bromion ved nukleofilt angrep av et bromidion eller et annet nukleofilt middel. Fra disse posisjonene bør det stereokjemiske resultatet være formasjonen transe-glykoler under nedbrytningen av sykliske epoksider. Faktisk, under den syrekatalyserte hydrolysen av cykloheksenoksid eller cyklopentenoksid, utelukkende transe-1,2-dioler.

Dermed tilsvarer to-trinns prosessen med alkenepoksidasjon etterfulgt av sur hydrolyse av epoksidet totalt reaksjonen anti-hydroksylering av alkener.
Begge stadier anti-Hydroksylering av alkener kan kombineres dersom alkenet behandles med vandig 30-70 % hydrogenperoksid i maur- eller trifluoreddiksyre. Begge disse syrene er sterke nok til å forårsake åpning av oksiranringen.

Basekatalysert åpning av oksiranringen fører også til dannelse av syklisk transe-glykoler.

Derfor er to-trinns prosessen med epoksidering av alkener etterfulgt av alkalisk hydrolyse av epoksider også en reaksjon anti-hydroksylering av alkener.
3. syn- Hydroksylering
Noen salter og oksider av overgangsmetaller i høyere oksidasjonstilstander er effektive reagenser syn-hydroksylering av dobbeltbindingen til en alken, når begge hydroksylgruppene legges til samme side av dobbeltbindingen. Oksidasjon av alkener med kaliumpermanganat er en av de eldste metodene syn Hydroksylering av en dobbeltbinding fortsetter å være mye brukt til tross for dens iboende begrensninger. Cis-1,2-cykloheksandiol ble først oppnådd av V.V. Markovnikov i 1878 ved hydroksylering av cykloheksen med en vandig løsning av kaliumpermanganat ved 0 0 C.

Denne metoden ble senere utviklet i verkene til den russiske forskeren E.E. Wagner altså syn-Hydroksylering av alkener under påvirkning av en vandig løsning av kaliumpermanganat kalles Wagner-reaksjonen. Kaliumpermanganat er et sterkt oksidasjonsmiddel som ikke bare kan hydroksylere dobbeltbindingen, men også spalte den resulterende vicinale diolen. For å unngå ytterligere nedbrytning av glykoler så mye som mulig, må reaksjonsbetingelsene kontrolleres nøye. Utbyttet av glykoler er vanligvis lave (30-60%). De beste resultatene oppnås ved hydroksylering av alkener i et lett alkalisk miljø (pH ~ 8 9) ved 0-5 0 C med en fortynnet 1 % vandig løsning av KMnO 4.


Til å begynne med produserer oksidasjonen av alkener med kaliumpermanganat en syklisk ester av mangansyre, som umiddelbart hydrolyseres til en vicinal diol.

Den sykliske esteren av mangansyre som et mellomprodukt ble ikke isolert, men dannelsen følger av eksperimenter med merket 18 O kaliumpermanganat: begge oksygenatomene i glykol viser seg å være merket under oksidasjonen av alkenet KMn 18 O 4. Dette betyr at begge oksygenatomene overføres fra oksidasjonsmidlet, og ikke fra løsningsmidlet - vann, som er i god overensstemmelse med den foreslåtte mekanismen.
En annen metode syn-hydroksylering av alkener under påvirkning av osmium (VIII) oksid OsO 4 ble foreslått av R. Kriege i 1936. Osmiumtetroksid er et fargeløst, flyktig, krystallinsk stoff, svært løselig i eter, dioksan, pyridin og andre organiske løsningsmidler. Når osmiumtetroksid reagerer med alkener i eter eller dioksan, dannes et svart bunnfall av syklisk osmisk syreester - osmat, som lett kan isoleres i individuell form. Tilsetningen av OsO 4 til dobbeltbindingen akselereres merkbart i oppløsning i pyridin. Dekomponeringen av osmater til vicinale glykoler oppnås ved virkningen av en vandig løsning av natriumhydrosulfitt eller hydrogensulfid.

Produktutbytte syn-Hydroksylering av alkener i denne metoden er betydelig høyere enn ved bruk av permanganat som oksidasjonsmiddel. En viktig fordel med Krige-metoden er fraværet av produkter av oksidativ spaltning av alkener, som er karakteristisk for permanganatoksidasjon.


Osmiumtetroksid er et veldig dyrt og vanskelig å få tak i, og det er også giftig. Derfor brukes osmium (VIII) oksid i syntesen av små mengder vanskelig tilgjengelige stoffer for å oppnå det høyeste utbyttet av diol. For å forenkle syn-hydroksylering av alkener under påvirkning av OsO 4, ble det utviklet en teknikk som tillater bruk av kun katalytiske mengder av dette reagenset. Hydroksylering av alkener utføres ved bruk av hydrogenperoksid i nærvær av OsO 4, for eksempel:

For å avslutte denne delen presenterer vi de stereokjemiske forholdene mellom alkenet cis- eller transe-konfigurasjon og konfigurasjonen av den resulterende vicinale diolen, som kan være cis- eller transe-isomer, erythro- eller trio-form, meso- eller D,L-form avhengig av substituentene i alkenet:

Lignende stereokjemiske forhold observeres i andre reaksjoner syn- eller anti- tilsetning ved flere bindinger av hydrogen, hydrogenhalogenider, vann, halogener, borhydrider og andre reagenser.